
New to Fiber-seq in the lab? Start with the Bench Protocol for the complete wet lab workflow from cells to sequencing-ready libraries.
This is also the book for the computational tools of Fiber-seq which describes the two major software tools:
fibertools(ft) which is a CLI tool for creating and interacting with Fiber-seq BAM files.- the
samtoolsof Fiber-seq
- the
- Fiber-seq Inferred Regulatory Elements which is a method for identifying regulatory elements on individual fibers and peak calling.
- the
MACS2of Fiber-seq
- the
Some Key features include:
- Predicting m6A sites from PacBio Fiber-seq data
- FIRE (Fiber-seq Inferred Regulatory Elements)
- Extracting Fiber-seq results into plain text files.
- Centering Fiber-seq results around a given position.
- pyft: Python bindings for
fibertools
A quick start guide can be found here.
You can help improve this book! And it is a easy as clicking the edit button in the top right.
Fiber-seq Bench Protocol
From nuclei to sequencing-ready libraries for PacBio HiFi and Oxford Nanopore platforms.
Fiber-seq uses the Hia5 N6-adenine methyltransferase to stencil chromatin architecture onto DNA in situ, enabling single-molecule, nucleotide-resolution readout of nucleosome positioning, chromatin accessibility, and transcription factor occupancy on multi-kilobase DNA fibers. This protocol covers the complete workflow from cell line harvest through sequencing-ready libraries for both PacBio HiFi and Oxford Nanopore platforms.
Part 1: Reagents, Buffers, and Equipment
Buffer A (Fiber-seq Reaction Buffer)
Larger stocks of Buffer A (e.g., 50-100 mL) can be made without spermidine, filter sterilized through a 0.22 um filter, and stored at room temperature safely for up to 6 months. Complete Buffer A can then be made by adding 1 uL of 0.5 M spermidine to 999 uL of the Buffer A stock immediately before use (see step 2.5).
| Component | Stock | Final | Per 1 mL |
|---|---|---|---|
| RNase/DNase-free H2O | 100% | --- | 960 uL |
| Tris-HCl, pH 8.0 | 1 M | 15 mM | 15 uL |
| NaCl | 5 M | 15 mM | 3 uL |
| KCl | 3 M | 60 mM | 20 uL |
| EDTA, pH 8.0 | 0.5 M | 1 mM | 2 uL |
| EGTA, pH 8.0 | 0.5 M | 0.5 mM | 1 uL |
| Spermidine | 0.5 M | 0.5 mM | 1 uL |
Note: We generally store 0.5 M spermidine at -20 C for several months. Aliquot into ~20 uL/tube. It is ok to freeze-thaw a few times.
2x Lysis Buffer (IGEPAL)
Buffer A supplemented with IGEPAL CA-630 (Sigma I8896) at a cell-type-specific concentration. The 2x stock is diluted 1:1 with the cell suspension during the lysis step. Prepare on ice.
| Cell Line | Final % IGEPAL | 2x Stock % | uL 10% IGEPAL per 1 mL Buffer A |
|---|---|---|---|
| K562 | 0.025 | 0.05 | 5 uL |
| HEK293 | 0.025 | 0.05 | 5 uL |
| HeLa | 0.05 | 0.1 | 10 uL |
| HepG2 | 0.05-0.075 | 0.1-0.15 | 10-15 uL |
| LCL (GM12878/GM24385) | 0.025-0.05 | 0.05-0.1 | 5-10 uL |
Note: For the lysis step, you can use either 0.025% or 0.05% final concentration of IGEPAL. Both do a good job isolating nuclei from multiple cell lines with minimal variability between the two concentrations. Generally, use 0.025% for more fragile cells (e.g., HEK293) and 0.05% for more robust ones. For retinal organoids or fibroblasts, Vollger et al. 2025 used 0.2% IGEPAL. For new cell lines, check lysis with trypan blue or AO/PI stain before proceeding.
Key Reagents
| Reagent | Source | Catalog # | Notes |
|---|---|---|---|
| Hia5 N6-adenine MTase | Lab-purified (primary) or EpiCypher CUTANA 15-1032 | --- | FPLC-purified, 100 U/uL, in 10% glycerol, store -80 C |
| SAM, 32 mM | NEB | B9003S | Stored at -20 C. SAM degrades over time; track expiration dates. Avoid excessive freeze-thaw by aliquoting. |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | NP-40 substitute. When making 10% IGEPAL, it takes time to dissolve. Heating to 37-40 C can help; we sometimes leave it on a rocker overnight. |
| Spermidine | Fisher Scientific | AC132740010 | Resuspend 1 g in 13.76 mL DNase/RNase-free water, filter sterilize with 0.22 um filter. Store 0.5 mL aliquots at -20 C. |
| SDS, 20% solution | Ambion/Invitrogen | AM9820 | For reaction quench |
| Qiagen MagAttract HMW DNA Kit | Qiagen | 67563 | Preferred extraction method |
| Promega Wizard HMW DNA Kit | Promega | A2920 | Alternative extraction method |
| PBS, 1x (no Ca/Mg) | Gibco | 10010023 | For wash and volume adjustment |
| Wide-bore pipette tips | Rainin | various | Essential for all DNA handling |
| DNA LoBind tubes | Eppendorf | 022431021 | 1.5 mL |
Equipment
- Refrigerated centrifuge (250-700 x g capable)
- Thermocycler with heated lid (25 C and 37 C)
- Qubit 4 fluorometer (Invitrogen)
Part 2: Cell Harvesting and Nuclei Isolation
Step 2.1 -- Harvest cells. Collect 1 x 10^6 cells per Fiber-seq reaction. Transfer from culture flask to a 1.5 mL LoBind tube. Pellet at 350 x g for 5 minutes. Remove supernatant completely.
Step 2.2 -- PBS wash. Resuspend in 1 mL PBS by gentle pipetting with a wide-bore tip. Pellet again at 350 x g for 5 minutes. Aspirate liquid completely.
Step 2.3 -- Resuspend in Buffer A. Gently resuspend the washed cell pellet in 60 uL of Buffer A by gently flicking the tube. Transfer the sample to a PCR tube using a wide-bore tip. Avoid generating bubbles.
Tip: Including 0.1% BSA in the PBS wash can help avoid sticky cell pellets.
Step 2.4 -- Lysis (nuclei isolation). Add 60 uL ice-cold 2x Lysis buffer (Buffer A + cell-type-specific IGEPAL; see table above). Gently resuspend using a mild finger flick. Do not vortex or pipette to resuspend. Incubate on ice for 10 minutes. The IGEPAL permeabilizes the plasma membrane while leaving nuclei and chromatin largely intact.
- Optionally, you can get a rough estimate of your lysis efficiency using a cell counter compatible with AO/PI staining. A complete lysis should have a viability count near 0%, with nuclei fluorescing red.
Critical: The detergent concentration is the single most important variable to optimize for new cell types. We find 0.025-0.05% final IGEPAL works well across most cell lines. If adapting to a new cell type, perform a lysis titration first.
Step 2.5 -- Pellet nuclei. Centrifuge at 350 x g for 5 minutes at 4 C. Carefully remove supernatant. The pellet should appear slightly translucent.
Step 2.6 -- Resuspend nuclei for methylation reaction. Resuspend the nuclei pellet in the complete reaction mix (see Part 3 table below). Proceed immediately to the Hia5 treatment.
Part 3: Hia5 Methyltransferase Treatment (In Situ m6A Labeling)
Hia5 is a nonspecific DNA N6-adenine methyltransferase (Drozdz et al., Nucleic Acids Research 2012; PMID: 22102579). It methylates adenines in accessible (non-nucleosome-bound) DNA while nucleosome-wrapped DNA is protected, creating a binary methylation stencil of chromatin architecture.
Step 3.1 -- Resuspend nuclei in reaction mix. Resuspend the nuclei pellet (from Step 2.5) directly in the complete reaction mix by gentle pipetting:
| Reagent | Volume |
|---|---|
| Buffer A | 57.5 uL |
| 32 mM SAM (NEB B9003S) | 1.5 uL |
| Hia5 MTase (200 U/uL, lab-purified) | 0.5 uL (100 U total) |
| Total reaction volume | 60 uL |
If using EpiCypher CUTANA Hia5 instead of lab-purified enzyme, adjust the volume to deliver 200 U total per million cells based on the lot-specific activity, and adjust the Buffer A volume to maintain 60 uL total.
Step 3.2 -- Mix and incubate. Gently flick the tube to resuspend nuclei in the reaction mix. Incubate on a heat block at 25 C for exactly 10 minutes. The 25 C temperature is critical for human cells. Do not exceed 10 minutes, as over-methylation reduces contrast between accessible and nucleosome-protected DNA.
Step 3.3 -- Quench the reaction. Add 3 uL of 20% SDS (final ~1% SDS). Gently invert the tube 10 times to mix. SDS denatures Hia5 and halts the reaction instantly.
Step 3.4 -- Bring to volume for gDNA extraction. Adjust volume for gDNA extraction per kit requirements. For Promega Wizard, add 37 uL for a final 100 uL and proceed. For Qiagen MagAttract add 117 uL ATL for a final 180 uL (see Part 4).
QC Checkpoint 1 -- Reaction parameters: Record exact incubation time, temperature, and Hia5 lot number/activity. The lab standard is 200 U Hia5 (at 200 U/uL) per million cells. Post-sequencing, the m6A/total adenines ratio should be 0.03-0.09 (median per molecule) or 5-7% globally. Under-methylation yields poor nucleosome resolution; over-methylation erases footprint signal.
Part 4: High Molecular Weight DNA Extraction
After quenching, the sample is a crude lysate of denatured chromatin in SDS. The goal is to extract intact, high molecular weight genomic DNA suitable for long-read sequencing. Bring the lysate volume up to the kit-required input with 1x PBS before proceeding.
Option A: Qiagen MagAttract HMW DNA Kit (Recommended)
Step 4A.1 -- Extract with Qiagen MagAttract HMW DNA Kit (cat# 67563). This bead-based method is the current lab recommendation. It is easier to use than column or precipitation methods and consistently yields more intact DNA. Bring the quenched lysate to 180 uL with ATL buffer, then follow the manufacturer's protocol. The MagAttract beads bind HMW DNA selectively. Elute in 100 uL Buffer AE. Incubate at 4 C overnight or over weekend. If yields are lower than expected, incubate tubes for 5 min at 56 C, shaking at 500 RPM.
Note: The Qiagen MagAttract kit has been much easier than the Promega kit so far, and generally yields more intact DNA. It also works well for primary tissue samples.
Option B: Promega Wizard HMW DNA Kit
Follow the manufacturer's protocol for the Promega Wizard HMW DNA Extraction Kit (cat# A2920).
Option C: Nanobind (PacBio) or NEB Monarch (Alternatives)
Other HMW extraction kits may also be used depending on lab availability.
Part 5: PacBio HiFi Library Preparation
This section follows the standard PacBio whole-genome HiFi library workflow using the SMRTbell prep kit 3.0 (PacBio cat# 102-182-700). Fiber-seq DNA is processed identically to standard WGS DNA. This is a PCR-free workflow throughout; amplification must never be used, as it would erase the m6A modifications.
5.1 DNA Shearing
Target fragment size: ~15-20 kb (optimal for HiFi CCS reads with >Q30 accuracy).
Option A (preferred): Megaruptor 3 (Diagenode)
- Dilute DNA to 83.3 ng/uL (5 ug in 60 uL)
- Run 2-cycle shearing method at speed 31-33
QC Checkpoint 3 -- Post-shearing: Femto Pulse or TapeStation. Mode should be 15-20 kb with minimal material <5 kb or >30 kb. Qubit quantify. Expect ~80% recovery.
Option B: Covaris g-TUBE (cat# 520079)
This variation is only recommended for very low input samples or for QC screening of deamination. Median read length will be ~10 kb.
- Load DNA (in 50-150 uL) into the g-TUBE
- For standard applications, follow the manufacturer's guidelines
- For low input (<100 ng), centrifuge in Eppendorf 5424R at 3,200 RPM for 2 minutes. Invert tube and repeat for a total of 4 passes. This step may require optimization depending on input gDNA fragment size, which can be checked by Femto Pulse.
- Recover sheared DNA
Notes for low input samples: It is relatively common for a small amount of liquid to be left on the top side of the column after a spin. If this occurs, add +1 min at 3,200 RPM, then +3x 30 sec at 3,400 RPM. Check after each additional spin; stop once fully through.
5.2 SMRTbell Library Construction (SPK 3.0)
Input: 1.25-5 ug sheared DNA. Follow the PacBio SMRTbell prep kit 3.0 protocol.
5.3 Sequencing
Step 5.3.1 -- Anneal-Bind-Cleanup. Follow the Revio SPRQ chemistry procedure. Anneal sequencing primer, bind SPRQ polymerase, perform final cleanup.
Step 5.3.2 -- Load. Load at 250 pM with adaptive loading onto a Revio SMRT Cell. Movie time: ~24 hours. Enable on-instrument 5mC and 6mA base modification calling (ICS v13.3+).
Expected output: 90-148 Gb HiFi data per SMRT Cell. At ~18 kb mean read length, this yields ~5-8 million HiFi reads, providing 26-30x genome coverage.
Part 6: Oxford Nanopore Ligation Library Preparation
An ONT-specific protocol is being finalized. In the meantime, follow the standard ONT LSK114 ligation sequencing protocol and use Dorado with the 6mA all-context model for basecalling.
Part 7: Quality Control and Expected Metrics
Pre-Sequencing QC Summary
| Checkpoint | Method | Acceptable Range |
|---|---|---|
| Cell count | Hemocytometer / counter | 1 x 10^6 per reaction (min) |
| HMW DNA yield | Qubit dsDNA HS | 3-6 ug per million LCL cells |
| DNA purity | NanoDrop | A260/280 = 1.8 +/- 0.1; A260/230 >= 2.0 |
| DNA integrity (pre-shear) | Femto Pulse / TapeStation | Mode >50 kb; GQN >= 7.0 at 10 kb |
| Post-shearing size | Femto Pulse / TapeStation | 15-20 kb (PacBio) or 20-35 kb (ONT) |
| SMRTbell library | Qubit | >= 300 ng total per Revio SPRQ SMRT Cell |
| ONT adapted library | Qubit | 200-700 ng; load ~300 ng |
Post-Sequencing QC (Fiber-seq-Specific)
Use the fibertools software suite to assess data quality:
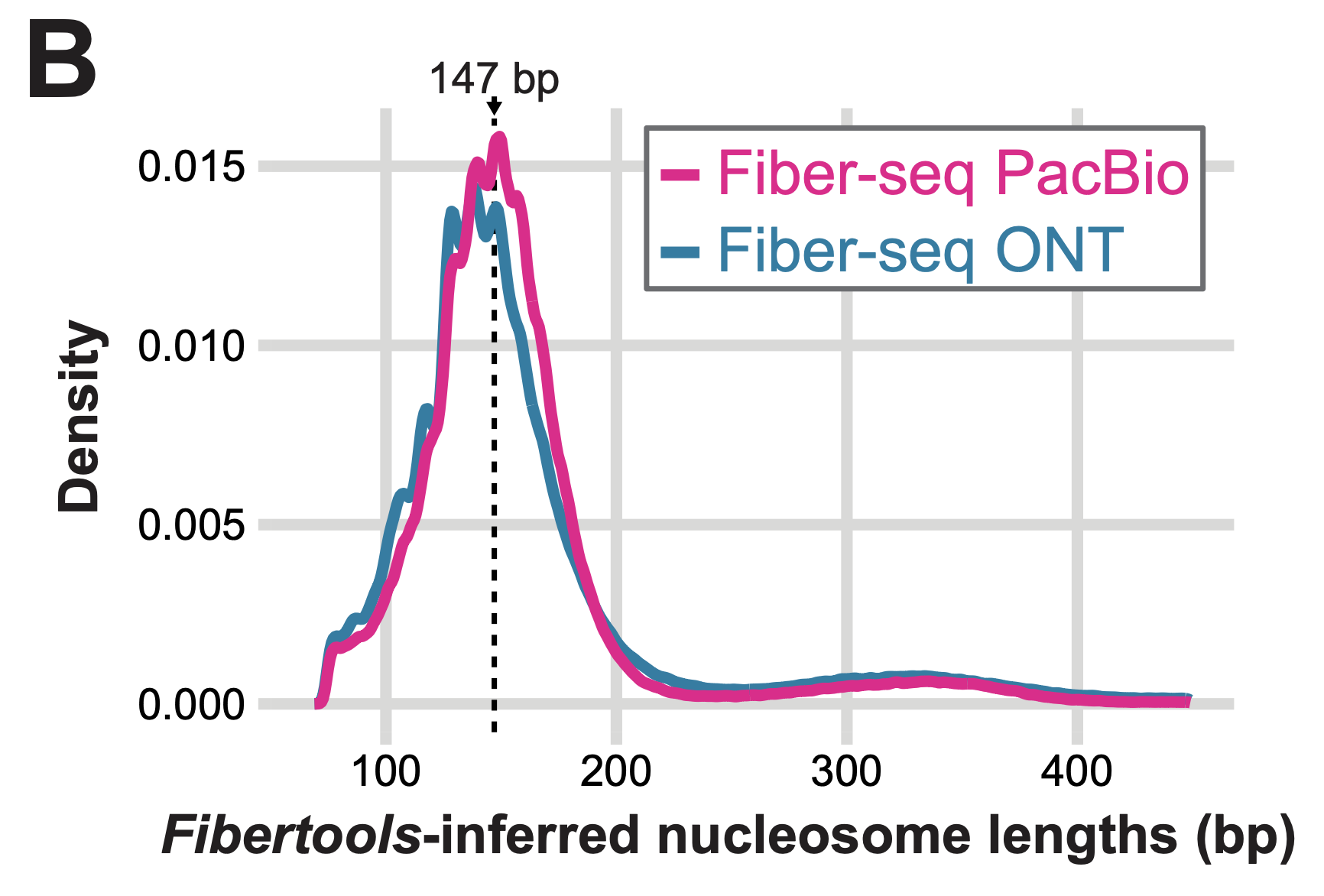
m6A autocorrelation function (ACF): Run ft qc --acf input.bam. A successful experiment produces a clear periodic signal at 147 bp, reflecting nucleosome protection. This is the single most important QC metric.
Global m6A fraction: Should be 0.03-0.09 (median per fiber) or 5-7% globally.
Nucleosome length distribution: After ft predict-m6a (PacBio) or ft add-nucleosomes (ONT/PacBio), footprint lengths should show a mode at ~147 bp.
FIRE element calling: Run the FIRE pipeline at >= 95% precision. FIRE peaks should overlap with DNase-HS and ATAC-seq peaks.
HiFi read metrics (PacBio): Read-length N50 of 17-18 kb, >93% bases at Q30+, >= 30x genome coverage.
Negative control: WGA DNA (no Hia5) processed identically should show <1% false-positive m6A.
Part 8: Workflow Summary and Timeline
| Day | Step | Time | Key Parameters |
|---|---|---|---|
| Day 1 | Cell harvest, PBS wash | 15 min | 1M LCL cells, 350 x g |
| Lysis | 10 min on ice | 0.025-0.05% IGEPAL final | |
| Hia5 labeling | 10 min at 25 C | 200 U Hia5, 0.8 mM SAM | |
| Quench + bring to volume | 2 min | 1% SDS final, PBS to kit volume | |
| HMW DNA extraction | 1-2 hr | Qiagen MagAttract (preferred) | |
| QC: Qubit + Femto Pulse | 30 min | Verify yield and size | |
| ~2-2.5 hr total from pellet to purified DNA | |||
| Day 2 (PacBio) | Shear DNA | 30 min | Megaruptor 3 or g-TUBE |
| SMRTbell library prep | 4-5 hr | SPK 3.0: Repair, Ligate, Nuclease | |
| Size selection | 1-2 hr | AMPure PB or BluePippin | |
| QC + ABC + Load Revio | ~3 hr | 250 pM, 24 hr movie | |
| Day 2 (ONT) | Optional shearing | 10 min | 26G needle, 25 passes |
| End-prep | 35 min | LSK114 + NEBNext E7672 | |
| Adapter ligation | 10-60 min | Room temperature | |
| QC + Load flow cell | 15 min | 300 ng on R10.4.1 |
Total hands-on time from cells to sequencing-ready library: ~6-8 hours (including QC pauses). The Hia5 labeling step itself takes only 10 minutes.
Essential Citations and Resources
Primary Fiber-seq Publications
- Stergachis AB et al. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 2020. 368(6498):1449-1454. DOI: 10.1126/science.aaz1646.
- Vollger MR et al. Synchronized long-read genome, methylome, epigenome, and transcriptome profiling resolve a Mendelian condition. Nature Genetics 2025. 57(2):469-479. DOI: 10.1038/s41588-024-02067-0.
- Jha A et al. DNA-m6A calling and integrated long-read epigenetic and genetic analysis with fibertools. Genome Research 2024. 34(11):1976-1986. DOI: 10.1101/gr.279095.124.
- Vollger MR et al. A haplotype-resolved view of human gene regulation. bioRxiv 2025. DOI: 10.1101/2024.06.14.599122. (FIRE pipeline).
- Bohaczuk SC et al. Resolving the chromatin impact of mosaic variants with targeted Fiber-seq. Genome Research 2024. 34(12):2269-2278.
- Peter CJ et al. Single chromatin fiber profiling and nucleosome position mapping in the human brain. Cell Reports Methods 2024. 4(12):100911.
- Swanson EG et al. Mapping single-cell diploid chromatin fiber architectures using DAF-seq. Nature Biotechnology 2025. DOI: 10.1038/s41587-025-02914-3.
- Drozdz M et al. Novel non-specific DNA adenine methyltransferases. Nucleic Acids Research 2012. 40(5):2119-2130. PMID: 22102579. (Hia5 enzyme characterization).
Online Protocols and Resources
- Protocols.io -- Brain Fiber-seq protocol (6mA labeling and HMW DNA extraction)
- EpiCypher CUTANA Hia5 (commercial Hia5 enzyme, SKU: 15-1032)
- EpiCypher CUTANA Fiber-seq Kit (complete kit with Hia5, SAM, nuclei extraction buffer)
- Fibertools documentation (computational guide for m6A calling and nucleosome annotation)
- fibertools-rs (GitHub) (CLI tool for Fiber-seq BAM processing)
- FIRE pipeline (GitHub) (Snakemake workflow for regulatory element calling)
- PacBio Fiber-seq Application Note (PacBio document 102-326-654 REV02, Oct 2025)
- ONT LSK114 Protocol (Ligation sequencing DNA V14)
- Dorado basecaller (use with 6mA all-context model for Fiber-seq)
The quick start guide for analyzing Fiber-seq data
The primary tool for handling Fiber-seq data is fibertools, and this page provides a high level order of operations for turning you raw Fiber-seq data into useful chromatin information. The steps differ slightly depending on if you are starting with PacBio or Oxford Nanopore Technologies (ONT); however, the steps can be summarized as:
- Create or filter m6A calls
- Infer nucleosomes and modification sensitive patches (MSPs)
- Apply the Fiber-seq QC tool
- Align and phase the data
- Validate your Fiber-seq BAM file
- Apply Fiber-seq inferred regulatory element (FIRE) calling to identify peaks and create UCSC browser tracks
Fiber-seq starting with PacBio
When using Fiber-seq data, it is important to check with your sequencing provider prior to sequencing to ensure that the output files contain the information required for Fiber-seq analysis.
Since 2025, Revio and Vega instruments automatically generate m6A calls on instrument using jasmine. For Revio,
automatic m6A calling was introduced with the SPRQ chemistry update; Revio runs since SPRQ include m6A calls.
Older Revio runs or runs from older instruments (Sequel II) require the CCS BAM output to include average kinetics information, which can be used to generate m6A calls off instrument. Without this information, Fiber-seq data cannot be analyzed.
Predict m6A and infer nucleosomes
Your PacBio data contains 6mA tags
As of 2025, PacBio's base modification caller jasmine automatically generates m6A predictions on instrument, in addition
to 5mC. You can check for the presence of these calls by inspecting the tags in your BAM file. m6A calls are encoded in
the MM tag and will appear with the prefix MM:Z:A+a, followed by positional information for the modified bases.
This removes the need to run ft predict and eliminates the requirement to retain polymerase kinetics tags in the PacBio
BAM file. However, you must still run ft add-nucleosomes, which is required for downstream Fiber-seq analysis.
ft add-nucleosomes -t 16 input.pacbio.bam output.fiberseq.bam
Your PacBio data does not have 6mA tags
Note:, the input CCS bam must have average kinetics to be able to call m6A.
To create useable Fiber-seq data you must first call m6A base-mods on the PacBio CCS bam using fibertools. First install fibertools and then process your bam file using the prediction command.
ft predict-m6a -t 16 input.ccs.bam output.fiberseq.bam
This will both make m6A calls and identify nucleosomes on each fiber.
Alignment and phasing
We recommend aligning with pbmm2 and phasing with HiPhase. Please see their respective documentation for more information.
Alternatively, we have written a snakemake pipeline to align and phase Fiber-seq data; however, this pipeline is not officially supported outside of our lab at this time.
After this point, you will have a Fiber-seq BAM file that is compatible with all the extraction commands in fibertools.
Validate your Fiber-seq BAM file
We have a quick validation tool which can test your BAM file for the desired Fiber-seq features. At this point you should have m6A calls, nucleosome calls, and aligned reads (phasing is optional).
ft validate output.fiberseq.bam
Total reads tested: 5000
Fraction with m6A: 100.00%
Fraction with nucleosomes: 100.00%
Number of FIRE calls: 0
Fraction aligned: 100.00%
Fraction phased: 85.10%
Fraction with kinetics: 0.00%
Fiber-seq peaks and UCSC browser tracks (FIRE)
Once you have a phased bam file, you can identify Fiber-seq inferred regulatory elements (FIREs) to call Fiber-seq peaks and make a UCSC trackHub.
You can find more details in on installing and running FIRE here: Running the FIRE pipeline.
Fiber-seq starting with Oxford Nanopore Technologies (ONT)
Predict m6A
ft predict-m6a does not include a model for ONT data; however, you can use software, such as Dorado, to add CpG and m6A to your ONT BAM file.
Alignment and phasing
You can either use Dorado to align your ONT data or use a tool like minimap2 to align your data. If you do use minimap2 be sure to include the flag -y to preserve the CpG and m6A information in the output BAM file.
If you do want to do phasing we recommend using WhatsHap for phasing ONT data. Please see their documentation for more information.
Filtering m6A calls
If you do use Dorado you must then filter the m6A calls with modkit using a tenth percentile cutoff for each flow-cell independently. This is the only way to get good m6A calls in our experience, and using any hard ML threshold will not hold between flow-cells. Here is an example command:
modkit call-mods -t 8 -p 0.1 input.dorado.bam filtered.dorado.bam
Infer nucleosomes and MSPs
Once you have CpG and m6A information in your filtered ONT BAM file, you can use ft add-nucleosomes to infer nucleosomes and MSPs.
ft add-nucleosomes filtered.dorado.bam output.bam
A full example for processing ONT data
Here is an example summary of the commands to process ONT data assuming you have already completed 6mA and CpG calling with dorado:
`#converts to fastq keeping all the BAM tags` \
samtools fastq -@ 8 -T "*" ONT.dorado.with.6mA.bam \
`#aligns the data inserting the tags back into the output BAM`
| minimap2 -t 32 --secondary=no -I 8G --eqx --MD -Y -y -ax map-ont reference.fasta - \
`#optionally add back in the read groups from the original bam using rustybam` \
| rb add-rg -u ONT.dorado.with.6mA.bam \
`#sort and index the BAM` \
| samtools sort -@ 32 --write-index -o tmp.ONT.fiberseq.bam
#filters the ONT data for the best 6mA calls and write to stdout
modkit call-mods -p 0.1 tmp.ONT.fiberseq.bam - \
`# adds nucleosome calls to the ONT Fiber-seq` \
| ft add-nucleosomes - ONT.fiberseq.bam
After this point, you will have a Fiber-seq BAM file that is compatible with all the extraction commands in fibertools.
Fiber-seq peaks and UCSC browser tracks (FIRE)
We have had good success in applying the FIRE pipeline to ONT data. However, this does require a heuristic in FIRE that must be enabled. To enable this add ont: true to your config.yaml file when setting up your FIRE run.
You can find more details in on installing and running FIRE here: Running the FIRE pipeline.

![]()




![]()
Think of fibertools as the samtools of Fiber-seq data. A command line tool for creating, filtering, interacting, and extracting from Fiber-seq BAM files.
This chapter covers:
- Creating a Fiber-seq BAM
- Extracting from a Fiber-seq BAM
- installing
fibertools - pyft: Python bindings
A complete list of subcommands and their help pages can be found here.
Creating a Fiber-seq bam file
This chapter covers creating a Fiber-seq bam file from a raw PacBio bam file. Including m6A prediction, nucleosome positioning, and FIRE identification.
ft predict-m6a
This command predicts m6A sites from PacBio Fiber-seq data. The input is a PacBio HiFi BAM file with kinetics and the output is a BAM file with the predicted m6A sites encoded in the ML and MM tags.
Details on the algorithm for m6A prediction are described in this publication: https://doi.org/10.1101/gr.279095.124.
ft add-nucleosomes
ft add-nucleosomes adds nucleosome positions and MSP positions to the Fiber-seq bam. The input is a Fiber-seq bam file with m6A calls and the output is a Fiber-seq bam file with the nucleosome positions encoded in the ns and nl tags and the MSP positions encoded in the as and al tags.
It is usually unnecessary to run this command manually, as it is called by ft predict-m6a automatically. However, you can run it manually if you want to adjust the parameters of the nucleosome calling algorithm.
Method
Nucleosome calling is performed by identifying stretches of DNA that are protected from Hia5 (i.e. do not have m6A signal). We have found the rate of false positive m6A calls in nucleosomes is very low when using fibertools (Jha et al.) allowing for a heuristic to perform as well as or better than our previous HMM caller (Dubocanin et al.).
There are three parameters in our heuristic nucleosome calling that can be adjusted: the minimum nucleosome length (n, default 75), the minimum combined nucleosome length (c, default 100), and the minimum extension to nucleosome length (e, default 25). These three parameters impact the three phases in nucleosome calling.
- Call all regions that have no m6A events for at least
nbases a candidate nucleosome. - Call all regions of size
cor more that have only one internal m6A (putative false positive) a candidate nucleosome. - Extend the length of nucleosomes identified in phases one and two if by spanning one additional m6A
ebases of unmodified sequence would be added to the nucleosome length.
ft fire
This command identifies Fiber-seq Inferred Regulatory Elements (FIREs) from a Fiber-seq BAM. The input is a Fiber-seq BAM file with m6A and nucleosome calls and the output is a Fiber-seq bam file with the FIREs encoded in the aq tags.
This command can be run in isolation; however, it is usually preferable to run the FIRE pipeline, which runs ft fire and performs many additional analyses and visualizations.
I just need the FIRE elements
If you only need FIRE elements in the BAM file and don't need the peak-calls or trackHubs that are part of the FIRE pipeline you can do a simplified run FIRE using only ft. e.g.:
# add FIREs to the BAM file
ft fire input.bam fire.bam
# Extract the FIREs from the BAM file into bed format
ft fire --extract fire.bam fire.bed.gz
# if you want the NUC calls and non-FIRE MSPs as well
ft fire --extract --all fire.bam all.bed.gz
Extracting from a fibertools bam
This chapter describes various subcommands that extract information from a fibertools bam file into plain text.
ft extract
Extract Fiber-seq data into plain text files
Inputs and options
See the help message for details.
Output description
All outputs to ft extract can be (and should be) compressed by simply adding the .gz extension.
For example, ft extract input.bam --m6a m6a.bed.gz will output a compressed bed12 file. Use - to output to stdout, e.g. ft extract input.bam --m6a -.
Shared Output columns:
| Column | Description |
|---|---|
| ct | Chromosome or contig |
| st | Start position of the read on the chromosome |
| en | End position of the read on the chromosome |
| fiber | The fiber/read name |
| score | The number of ccs passes for the read (rounded) |
| strand | The strand of the read alignment |
Columns specific to the --m6a, --cpg, --nuc, and --msp outputs
All of these files are written in standard bed12 format. The first and last block in each the bed12 record do not reflect real data, and exist only to mark the start and end positions of the read. If you would like to convert these beds into bigBeds be sure to include -allow1bpOverlap in your command.
| Column | Description |
|---|---|
| thick start | Same as the start (st) |
| thick end | Same as the end (en) |
| itemRgb | Color specifc to the datatype, e.g. m6a marks get a purple RGB |
| blockCount | The number of blocks in the bed12 record |
| blockSizes | A comma separated list of the lengths of each feature in the bed12 record |
| blockStarts | A comma separated list of the relative start positions of each block in the bed12 record |
Columns specific to the --all output
| Column | Description |
|---|---|
| sam_flag | The sam flag of the read alignment |
| HP | The haplotype tag for the read |
| RG | The read group tag for the read |
| fiber_length | The length of the read in bp |
| fiber_sequence | The sequence of the read |
| ec | The number of ccs passes for the read (no rounding) |
| rq | The estimated accuracy of the read |
| total_AT_bp | The total number of AT bp in the read |
| total_m6a_bp | The total number of m6a bp in the read |
| total_nuc_bp | The total number of nucleosome bp in the read |
| total_msp_bp | The total number of MSP bp in the read |
| total_5mC_bp | The total number of 5mC bp in the read |
| nuc_starts | The start positions of the nucleosomes in molecular coordinates (comma separated list) |
| nuc_lengths | The lengths of the nucleosomes in molecular coordinates (comma separated list) |
| ref_nuc_starts | The start positions of the nucleosomes in reference coordinates (comma separated list) |
| ref_nuc_lengths | The lengths of the nucleosomes in reference coordinates (comma separated list) |
| msp_starts | The start positions of the MSPs in molecular coordinates (comma separated list) |
| msp_lengths | The lengths of the MSPs in molecular coordinates (comma separated list) |
| fire | The quality score of the MSP as a FIRE element (if FIRE as been applied). Scores over 230 are FIRE elements (comma separated list, range 0-255) |
| ref_msp_starts | The start positions of the MSPs in reference coordinates (comma separated list) |
| ref_msp_lengths | The lengths of the MSPs in reference coordinates (comma separated list) |
| m6a | The start positions of the m6a in molecular coordinates (comma separated list) |
| ref_m6a | The start positions of the m6a in reference coordinates (comma separated list) |
| m6a_qual | The quality of the m6a positions (ML value, comma separated list) |
| 5mC | The start positions of the 5mC in molecular coordinates (comma separated list) |
| ref_5mC | The start positions of the 5mC in reference coordinates (comma separated list) |
| 5mC_qual | The quality of the 5mC positions (ML value, comma separated list) |
Note positions in columns starting with ref_ maybe contain -1 (NA) values if the reference sequence has an insertion or deletion relative to the read sequence at that position.
ft center
This command centers Fiber-seq data around given reference positions. This is useful for making aggregate m6A and CpG observations, as well as visualization of SVs
Inputs and options
See the help message for details.
Output description
This command writes Fiber-seq data in a tab-delimited format to stdout that has been centered relative to positions specified in the input bed file.
| Column | Description |
|---|---|
| chrom | Chromosome |
| centering_position | The position on the chromosome about which data is being centered. |
| strand | The strand of the centering position. |
| subset_sequence | The sequence of the read around the centering position. |
| reference_start | The start position of the read in the reference. |
| reference_end | The end position of the read in the reference. |
| query_name | The name of the sequencing read |
| RG | The read group the read belongs to |
| centered_query_start | The start position of the read relative to the centering position |
| centered_query_end | The end position of the read relative to the centering position |
| query_length | The length of the read |
Additional columns specific to long (default) format
| Column | Description |
|---|---|
| centered_position_type | The type of position being centered. One of: m6A, 5mC, nuc, msp. |
| centered_start | The start position of the "feature" relative to the centering position |
| centered_end | The end position of the "feature" relative to the centering position |
| centered_quality | The quality of the "feature" relative to the centering position (ML value) |
Additional columns specific to the wide format
| Column | Description |
|---|---|
| centered_m6a_positions | A comma separated list of m6a positions |
| m6a_qual | The quality of the m6a positions (ML value) |
| centered_5mC_positions | A comma separated list of 5mC positions |
| 5mC_qual | The quality of the 5mC positions (ML value) |
| centered_nuc_starts | A comma separated list of nuc starts |
| centered_nuc_ends | A comma separated list of nuc ends |
| centered_msp_starts | A comma separated list of msp starts |
| centered_msp_ends | A comma separated list of msp ends |
| query_sequence | The sequence of the read |
Note if the --wide flag is used with the --reference flag some positions in the comma separated lists can be NA when the reference sequence has an insertion or deletion relative to the read sequence at that position.
ft footprint
Usage
ft footprint [OPTIONS] <BAM> --bed <BED> --yaml <YAML>
The BAM file is an indexed fiber-seq bam file.
The BED file is a bed file with the motifs you'd like to test for footprints. This should include the strand the motif is on.
The YAML file is a file that describes the modules within the motif that can be footprinted. e.g. a CTCF yaml with its multiple binding sites might look like:
modules:
- [0, 8]
- [8, 16]
- [16, 23]
- [23, 29]
- [29, 35]
Modules must start at zero, end at the length of the motif, be sorted, and be contiguous with one another. At most 15 modules are allowed, and the intervals are 0-based, half-open (like BED).
See the help message for details.
Description of output columns
The footprinting output table is a tab-separated file with the same number of entries as the input BED file and the following columns:
| Column | Description |
|---|---|
| chrom | Chromosome |
| start | The start position of the motif |
| end | The end position of the motif |
| strand | The strand of the motif. |
| n_spanning_fibers | The number of fibers that span the motif. |
| n_spanning_msps | The number of msp that span the motif. |
| n_overlapping_nucs | The number of fibers that have an intersecting nucleosome. |
| module_X | The number of fibers that are footprinted in module X. The number of module columns is determined by the footprinting yaml. |
| footprint_codes | Comma separated list of footprint codes for each fiber. See details below. |
| fire_quals | Comma separated list of fire qualities for each fiber. -1 if the MSP is not spanning or present. Note all fire_quals will be 0 or -1 if FIRE has not been applied to the bam. |
| fiber_names | Comma separated list of fiber names that span the motif. Names share the same index as the previous column, so they can be matched with footprint codes. |
Footprint codes
The footprint codes are an encoded bit flag similar to how filtering is done with samtools. If the first bit is set (1) then all modules within the motif are fully overlapped by an MSP on that fiber. For each following bit, the bit is set if that module does not contain m6A events, presumably due to transcription factor or nucleosome binding across the module on that fiber.
Here are some examples in python for how you could test a footprint code in a few ways:
fp_code = 0b1001 # this is a value of 9, but in binary it is 1001
# test if the first bit is set, there is a spanning MSP, true in this example
(fp_code & 1) > 0
# test if the first module is footprinted, false in this example
(fp_code & (1 << 1)) > 0
# test if the third module is footprinted, true in this example
(fp_code & (1 << 3)) > 0
ft-pileup
The ft-pileup command is used to generate a per-base pileup of Fiber-seq data. This command is useful for visualizing the distribution of various features across the genome.
Inputs and options
See the help message for details.
ft-qc
The ft-qc command is used to generate quality control metrics for a Fiber-seq BAM file.
Inputs and options
See the help message for details.
Help pages for fibertools subcommands
ft
Fiber-seq toolkit in rust
Usage: ft [OPTIONS] <COMMAND>
Commands:
predict-m6a Predict m6A positions using HiFi kinetics data and encode the
results in the MM and ML bam tags. Also adds nucleosome (nl, ns) and
MTase sensitive patches (al, as) [aliases: m6A, m6a]
add-nucleosomes Add nucleosomes to a bam file with m6a predictions
fire Add FIREs (Fiber-seq Inferred Regulatory Elements) to a bam file
with m6a predictions
extract Extract fiberseq data into plain text files [aliases: ex, e]
center This command centers fiberseq data around given reference positions.
This is useful for making aggregate m6A and CpG observations, as
well as visualization of SVs [aliases: c, ct]
footprint Infer footprints from fiberseq data
qc Collect QC metrics from a fiberseq bam file
track-decorators Make decorated bed files for fiberseq data
pileup Make a pileup track of Fiber-seq features from a FIRE bam
clear-kinetics Remove HiFi kinetics tags from the input bam file
strip-basemods Strip out select base modifications
ddda-to-m6a Convert a DddA BAM file to pseudo m6A BAM file
fiber-hmm Apply FiberHMM to a bam file
validate Validate a Fiber-seq BAM file for m6A, nucleosome, and optionally
FIRE calls
pg-inject Create a mock BAM file from a reference FASTA with perfectly aligned
sequences
pg-lift Lift annotations through a pangenome graph from source to target
coordinates
pg-pansn Add or strip panSN-spec prefixes from BAM contig names
call-peaks Call FIRE peaks using FDR-based peak calling on pileup data
[aliases: peaks, call]
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft predict-m6a
Predict m6A positions using HiFi kinetics data and encode the results in the MM and ML
bam tags. Also adds nucleosome (nl, ns) and MTase sensitive patches (al, as)
Usage: ft predict-m6a [OPTIONS] [BAM] [OUT]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
[OUT] Output bam file with m6A calls in new/extended MM and ML bam tags [default: -]
Options:
-n, --nucleosome-length <NUCLEOSOME_LENGTH>
Minium nucleosome length [default: 75]
-c, --combined-nucleosome-length <COMBINED_NUCLEOSOME_LENGTH>
Minium nucleosome length when combining over a single m6A [default: 100]
--min-distance-added <MIN_DISTANCE_ADDED>
Minium distance needed to add to an already existing nuc by crossing an m6a
[default: 25]
-d, --distance-from-end <DISTANCE_FROM_END>
Minimum distance from the end of a fiber to call a nucleosome or MSP [default:
45]
-k, --keep
Keep hifi kinetics data
-h, --help
Print help (see more with '--help')
-V, --version
Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
Developer-Options:
--force-min-ml-score <FORCE_MIN_ML_SCORE>
Force a different minimum ML score
--all-calls
Keep all m6A calls regardless of how low the ML value is
-b, --batch-size <BATCH_SIZE>
Number of reads to include in batch prediction [default: 1]
ft fire
ft extract
Extract fiberseq data into plain text files
Usage: ft extract [OPTIONS] [BAM]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
Options:
-r, --reference Report positions in reference sequence coordinates
--molecular Report positions in the molecular sequence coordinates
--m6a <M6A> Output path for m6a bed12
-c, --cpg <CPG> Output path for 5mC (CpG, primrose) bed12
--msp <MSP> Output path for methylation sensitive patch (msp) bed12
-n, --nuc <NUC> Output path for nucleosome bed12
-a, --all <ALL> Output path for a tabular format including "all" fiberseq information
in the bam
-h, --help Print help (see more with '--help')
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
All-Format-Options:
-q, --quality Include per base quality scores in "fiber_qual"
-s, --simplify Simplify output by removing fiber sequence
ft center
This command centers fiberseq data around given reference positions. This is useful for
making aggregate m6A and CpG observations, as well as visualization of SVs
Usage: ft center [OPTIONS] --bed <BED> [BAM]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
Options:
-b, --bed <BED> Bed file on which to center fiberseq reads. Data is adjusted to the
start position of the bed file and corrected for strand if the
strand is indicated in the 6th column of the bed file. The 4th
column will also be checked for the strand but only after the 6th
is. If you include strand information in the 4th (or 6th) column it
will orient data accordingly and use the end position of bed record
instead of the start if on the minus strand. This means that
profiles of motifs in both the forward and minus orientation will
align to the same central position
-d, --dist <DIST> Set a maximum distance from the start of the motif to keep a
feature
-w, --wide Provide data in wide format, one row per read
-r, --reference Return relative reference position instead of relative molecular
position
-s, --simplify Replace the sequence output column with just "N"
-h, --help Print help (see more with '--help')
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft add-nucleosomes
Add nucleosomes to a bam file with m6a predictions
Usage: ft add-nucleosomes [OPTIONS] [BAM] [OUT]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
[OUT] Output bam file with nucleosome calls [default: -]
Options:
-n, --nucleosome-length <NUCLEOSOME_LENGTH>
Minium nucleosome length [default: 75]
-c, --combined-nucleosome-length <COMBINED_NUCLEOSOME_LENGTH>
Minium nucleosome length when combining over a single m6A [default: 100]
--min-distance-added <MIN_DISTANCE_ADDED>
Minium distance needed to add to an already existing nuc by crossing an m6a
[default: 25]
-d, --distance-from-end <DISTANCE_FROM_END>
Minimum distance from the end of a fiber to call a nucleosome or MSP [default:
45]
-h, --help
Print help
-V, --version
Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft footprint
Infer footprints from fiberseq data
Usage: ft footprint [OPTIONS] --bed <BED> --yaml <YAML> [BAM]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
Options:
-b, --bed <BED> BED file with the regions to footprint. Should all contain the same
motif with proper strand information, and ideally be ChIP-seq peaks
-y, --yaml <YAML> yaml describing the modules of the footprint
-o, --out <OUT> Output bam [default: -]
-h, --help Print help
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft clear-kinetics
Remove HiFi kinetics tags from the input bam file
Usage: ft clear-kinetics [OPTIONS] [BAM] [OUT]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
[OUT] Output bam file without hifi kinetics [default: -]
Options:
-h, --help Print help
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft strip-basemods
Strip out select base modifications
Usage: ft strip-basemods [OPTIONS] [BAM] [OUT]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
[OUT] Output bam file [default: -]
Options:
-b, --basemod <BASEMOD> base modification to strip out of the bam file [possible
values: m6A, 6mA, 5mC, CpG]
--ml-m6a <ML_M6A> filter out m6A modifications with less than this ML value
[default: 0]
--ml-5mc <ML_5MC> filter out 5mC modifications with less than this ML value
[default: 0]
--drop-forward Drop forward strand of base modifications
--drop-reverse Drop reverse strand of base modifications
-h, --help Print help
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft track-decorators
Make decorated bed files for fiberseq data
Usage: ft track-decorators [OPTIONS] --bed12 <BED12> [BAM]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
Options:
-b, --bed12 <BED12> Output path for bed12 file to be decorated
-d, --decorator <DECORATOR> Output path for decorator bed file [default: -]
-h, --help Print help
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft pileup
Make a pileup track of Fiber-seq features from a FIRE bam
Usage: ft pileup [OPTIONS] [BAM] [RGN]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
[RGN] Region string to make a pileup of. e.g. chr1:1-1000 or chr1:1-1,000 If not
provided will make a pileup of the whole genome
Options:
-o, --out <OUT> Output file [default: -]
-m, --m6a include m6A calls
-c, --cpg include 5mC calls
--haps For each column add two new columns with the hap1 and
hap2 specific data
-k, --keep-zeros Keep zero coverage regions
-p, --per-base Write output one base at a time even if the values do
not change
--fiber-coverage Calculate coverage starting from the first MSP/NUC to
the last MSP/NUC position instead of the complete
span of the read alignment
--shuffle <SHUFFLE> Shuffle the fiber-seq data according to a bed file of
the shuffled positions of the fiber-seq data
--rolling-max <ROLLING_MAX> Output a rolling max of the score column over X bases
--no-msp No MSP columns
--no-nuc No NUC columns
-h, --help Print help (see more with '--help')
-V, --version Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
ft qc
Collect QC metrics from a fiberseq bam file
Usage: ft qc [OPTIONS] [BAM] [OUT]
Arguments:
[BAM] Input BAM file. If no path is provided stdin is used. For m6A prediction, this
should be a HiFi bam file with kinetics data. For other commands, this should
be a bam file with m6A calls [default: -]
[OUT] Output text file with QC metrics. The format is a tab-separated file with the
following columns: "statistic\tvalue\tcount" where "statistic" is the name of
the metric, "value" is the value of the metric, and "count" is the number of
times the metric was observed [default: -]
Options:
--acf
Calculate the auto-correlation function of the m6A marks in the fiber-seq data
--acf-max-lag <ACF_MAX_LAG>
maximum lag for the ACF calculation [default: 250]
--acf-min-m6a <ACF_MIN_M6A>
Minimum number of m6A marks to use a read in the ACF calculation [default:
100]
--acf-max-reads <ACF_MAX_READS>
maximum number of reads to use in the ACF calculation [default: 10000]
--acf-sample-rate <ACF_SAMPLE_RATE>
After sampling the first "acf-max-reads" randomly sample one of every
"acf-sample-rate" reads and replace one of the previous reads at random
[default: 100]
-m, --m6a-per-msp
In the output include a measure of the number of m6A events per MSPs of a
given size. The output format is: "m6a_per_msp_size\t{m6A count},{MSP
size},{is a FIRE}\t{count}" e.g. "m6a_per_msp_size\t35,100,false\t100"
--n-reads <N_READS>
Only process the first "n" reads in the input bam file
-h, --help
Print help
-V, --version
Print version
BAM-Options:
-F, --filter <BIT_FLAG> BAM bit flags to filter on, equivalent to `-F` in
samtools view For call-peaks: defaults to 2304 (filters
secondary and supplementary alignments) For other
commands: defaults to 0 (no filtering)
-x, --ftx <FILTER_EXPRESSION> Filtering expression to use for filtering records
Example: filter to nucleosomes with lengths greater
than 150 bp -x "len(nuc)>150" Example: filter to msps
with lengths between 30 and 49 bp -x "len(msp)=30:50"
Example: combine 2+ filter expressions -x
"len(nuc)<150,len(msp)=30:50" Filtering expressions
support len() and qual() functions over msp, nuc, m6a,
cpg
--ml <MIN_ML_SCORE> Minium score in the ML tag to use or include in the
output [env: FT_MIN_ML_SCORE=] [default: 125]
-u, --uncompressed Output uncompressed BAM files
Global-Options:
-t, --threads <THREADS> Threads [default: 8]
Debug-Options:
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace]
--quiet Turn off all logging
pyft: Python bindings for fibertools
![]()
See the documentation for pyft on readthedocs for more information.
Installation
This chapter covers the various ways to install fibertools.
It is easiest to install fibertools using conda (instructions). However, for the fastest m6A predictions you need to install with libtorch enabled (instructions) if you would like to install from source, you can use the following instructions.
From bioconda
fibertools-rs is avalible through bioconda and can be installed with the following command:
mamba install -c conda-forge -c bioconda fibertools-rs
However, due to size constraints in bioconda this version does not support contain the pytorch libraries or GPU acceleration for m6A predictions. m6A predictions will still work in the bioconda version but may be much slower. If you would like to use m6A prediction and GPU acceleration, you will need to install using the directions here.
From crates.io
Installation from crates.io requires the rust package manager cargo. You can find how to install cargo here.. Furthermore, a recent version of gcc and cmake is required. I have tested and recommend gcc v10.2.0 and cmake v3.21.1, though other versions may work.
cargo install fibertools-rs
From GitHub (active development)
Using cargo from source:
cargo install --git https://github.com/fiberseq/fibertools-rs
or using git form source:
git clone https://github.com/fiberseq/fibertools-rs
cd fibertools-rs
cargo build --release
./target/release/ft --help
With libtorch
Get libtorch v2.2.0 from the PyTorch website and extract the content of the zip file.
- On Linux/Unix system you can download with:
wget https://download.pytorch.org/libtorch/cu118/libtorch-shared-with-deps-2.2.0%2Bcu118.zip
- On macOS you can download with:
wget https://download.pytorch.org/libtorch/cpu/libtorch-macos-2.2.0.zip
- Windows is not supported and will not be.
Then add the following to your .bashrc or equivalent, where /path/to/libtorch is the path to the directory that was created when unzipping the file:
export LIBTORCH_CXX11_ABI=0
export LIBTORCH=/path/to/libtorch # e.g. export LIBTORCH=/Users/mrvollger/lib/libtorch
export LD_LIBRARY_PATH=${LIBTORCH}/lib:$LD_LIBRARY_PATH
export DYLD_LIBRARY_PATH=${LIBTORCH}/lib:$LD_LIBRARY_PATH
Finally install using cargo:
cargo install --all-features fibertools-rs
FiberHMM: HMM-based chromatin footprint calling


FiberHMM is a hidden Markov model toolkit for calling chromatin footprints and methylase-sensitive patches (MSPs) from single-molecule data. It works with Fiber-seq (m6A via Hia5) and DAF-seq (deamination via DddA/DddB), producing fibertools-compatible BAM output with ns/nl/as/al tags.
By learning sequence-context-specific emission probabilities from control data, FiberHMM accounts for the large biases in methylation efficiency across different hexamer contexts. This is what enables accurate footprint calling at much smaller sizes than heuristic approaches -- resolving not just nucleosomes but also transcription factor binding sites, Pol II, and other sub-nucleosomal footprints that would otherwise be lost in context noise.
FiberHMM also provides a unified framework for calling footprints across different single-molecule chemistries (Hia5, DddA, DddB) and sequencing platforms (PacBio, Nanopore), making it straightforward to directly compare chromatin accessibility measurements between experiments.
This chapter covers:
The source code and full documentation can be found on GitHub.
Installation and usage
Install
pip install fiberhmm
Or from source:
git clone https://github.com/fiberseq/FiberHMM.git
cd FiberHMM
pip install -e .
Optional: install numba for ~10x faster HMM computation, and matplotlib for --stats diagnostic plots.
pip install numba matplotlib
Pipeline overview
FiberHMM has four main steps, each with a dedicated command:
- Generate emission probabilities from control data (
fiberhmm-probs) - Train the HMM on your sample (
fiberhmm-train) - Call footprints on your experiment (
fiberhmm-apply) - Extract to BED12/bigBed for visualization (
fiberhmm-extract)
If you already have a pre-trained model for your chemistry, you can skip directly to step 3.
Generating emission probabilities
This requires two control BAMs: one with accessible (naked/dechromatinized) DNA and one with inaccessible (native chromatin) DNA. These define how methylation rates differ between accessible and inaccessible states for each sequence context.
fiberhmm-probs \
-a accessible_control.bam \
-u inaccessible_control.bam \
-o probs/ \
--mode pacbio-fiber \
--stats
This produces emission probability tables in probs/tables/ for each context size.
Training the HMM
fiberhmm-train \
-i sample.bam \
-p probs/tables/accessible_A_k3.tsv probs/tables/inaccessible_A_k3.tsv \
-o models/ \
-k 3 \
--stats
Output: models/best-model.json (the trained model).
Calling footprints
fiberhmm-apply \
-i experiment.bam \
-m models/best-model.json \
-o output/ \
-c 8 \
--scores
This writes a BAM file with fibertools-compatible footprint tags:
| Tag | Type | Description |
|---|---|---|
ns | B,I | Footprint starts (0-based query coords) |
nl | B,I | Footprint lengths |
as | B,I | MSP starts |
al | B,I | MSP lengths |
nq | B,C | Footprint quality scores (0-255, with --scores) |
aq | B,C | MSP quality scores (0-255, with --scores) |
The output BAM is compatible with ft extract, FIRE, and any other tool that reads fibertools-style tags.
Extracting to BED12/bigBed
fiberhmm-extract -i output/experiment_footprints.bam
This produces bigBed files for footprints, MSPs, m6A, and m5C that can be loaded into genome browsers.
Analysis modes
| Mode | Flag | Chemistry | Target bases |
|---|---|---|---|
| PacBio fiber-seq | --mode pacbio-fiber | Hia5 m6A, both strands | A, T (with RC) |
| Nanopore fiber-seq | --mode nanopore-fiber | Hia5 m6A, single strand | A only |
| DAF-seq | --mode daf | DddA/DddB deamination | C or G |
The pacbio-fiber vs nanopore-fiber distinction only matters for Hia5, where PacBio detects modifications on both strands while Nanopore detects only one. For deaminase-based methods (DddA, DddB), --mode daf is always used regardless of sequencing platform.
Utilities
fiberhmm-utils provides additional commands for model management:
# Convert legacy models to JSON
fiberhmm-utils convert old_model.pickle new_model.json
# Inspect model parameters
fiberhmm-utils inspect model.json
# Transfer emission probs between chemistries
fiberhmm-utils transfer --target daf.bam --reference-bam fiber.bam -o probs/ --mode daf
# Scale emission probabilities
fiberhmm-utils adjust model.json --state accessible --scale 1.1 -o adjusted.json
Performance tips
- Use
-c 8(or more cores) for parallel footprint calling - For small genomes, reduce
--region-size(500KB for yeast, 2MB for Drosophila) - Install
numbafor faster HMM training - Use
--skip-scaffoldsto avoid processing thousands of contigs
Pre-trained models
FiberHMM ships with pre-trained models in the models/ directory. These can be used directly with fiberhmm-apply without needing to generate emission probabilities or train from scratch.
Available models
| Model | File | Enzyme | Platform | Mode | Organism |
|---|---|---|---|---|---|
| Hia5 PacBio | hia5_pacbio.json | Hia5 (m6A) | PacBio | pacbio-fiber | Drosophila |
| Hia5 Nanopore | hia5_nanopore.json | Hia5 (m6A) | Nanopore | nanopore-fiber | Yeast |
| DddA PacBio | ddda_pacbio.json | DddA (deamination) | PacBio | daf | Drosophila |
| DddB Nanopore | dddb_nanopore.json | DddB (deamination) | Nanopore | daf | Drosophila |
Using a pre-trained model
# PacBio Hia5 fiber-seq
fiberhmm-apply -i experiment.bam -m models/hia5_pacbio.json -o output/ -c 8
# Nanopore Hia5 fiber-seq
fiberhmm-apply -i experiment.bam -m models/hia5_nanopore.json -o output/ -c 8
# DAF-seq (DddA or DddB)
fiberhmm-apply -i experiment.bam -m models/ddda_pacbio.json -o output/ -c 8 --mode daf
The mode and context size are stored in the model JSON and auto-detected at runtime, so you generally do not need to specify --mode or -k when using a pre-trained model.
When to train your own model
The pre-trained models work well as a starting point, but you may want to train a custom model if:
- You are working with a different organism where chromatin structure differs substantially
- You are using a different methyltransferase or deaminase chemistry
- You have matched accessible and inaccessible controls for your specific experiment
See the usage guide for the full training pipeline.
Inspecting a model
You can inspect any model's parameters with:
fiberhmm-utils inspect models/hia5_pacbio.json
This prints the mode, context size, start probabilities, transition matrix, and emission probability summary statistics.
How FiberHMM works
The problem
Single-molecule chromatin assays (Fiber-seq, DAF-seq) mark accessible DNA with enzymatic modifications -- m6A methylation or cytosine deamination. But the efficiency of these enzymes depends on the local sequence context: some hexamers are methylated much more readily than others, even in fully accessible DNA. Without accounting for this, a naive threshold would over-call footprints in low-efficiency contexts and under-call them in high-efficiency ones.
The approach
FiberHMM uses a two-state hidden Markov model where:
- State 0 (inaccessible): The DNA is nucleosome-protected or otherwise inaccessible. Modification rates are low.
- State 1 (accessible): The DNA is in a methylase-sensitive patch (MSP). Modification rates are high.
The key insight is that emission probabilities are learned per sequence context (hexamer) from control data, so the model knows the expected modification rate for each context in each state. This allows it to correctly interpret a low modification rate at a context that is inherently hard to methylate versus a genuinely protected region.
Pipeline steps
1. Emission probability estimation
Control BAMs define how modification rates differ between accessible and inaccessible states:
- Accessible control (e.g., naked DNA treated with methyltransferase): Gives
P(modified | accessible, context)for each hexamer - Inaccessible control (e.g., native chromatin): Gives
P(modified | inaccessible, context)for each hexamer
These context-specific emission probabilities are stored as TSV tables, one per context size k.
2. Training
The Baum-Welch (EM) algorithm learns the transition probabilities and start probabilities from sample data, while the emission probabilities remain fixed from the control data. Multiple random initializations are used to avoid local optima, and the model with the best log-likelihood is selected.
3. Decoding
The Viterbi algorithm finds the most likely state sequence for each read. Contiguous runs of state 0 become footprints; contiguous runs of state 1 become MSPs. Optionally, the forward-backward algorithm computes posterior probabilities for per-footprint confidence scores.
Sequence context encoding
Each position on a read is encoded as a hexamer centered on the target base (for k=3, a 7-mer). The hexamer is mapped to an integer code using a deterministic lookup table. For PacBio fiber-seq, reverse complement contexts are merged to account for double-stranded m6A detection. The total number of symbols is 4^(2k) (or 4^(2k) * 2 with separate modified/unmodified codes).
Context is computed directly from the read sequence -- no genome reference or context files are needed.
Why context matters for small footprints
Nucleosome-scale footprints (~150 bp) are large enough that context biases average out over the footprint length. But for smaller features -- transcription factor binding sites (~10-30 bp), Pol II (~50 bp) -- individual hexamer biases dominate the signal. A 20 bp stretch of low-efficiency contexts can look identical to a genuine TF footprint if context isn't modeled.
By learning the expected modification rate for each hexamer in each chromatin state, FiberHMM can distinguish true small footprints from context artifacts, enabling accurate sub-nucleosomal footprint calling.
FIRE: descriptions, methods, and outputs
The code for running the FIRE pipeline can be found on GitHub, and if you do use FIRE in your work please cite our publication:
Vollger, M. R., Swanson, E. G., Neph, S. J., Ranchalis, J., Munson, K. M., Ho, C.-H., Sedeño-Cortés, A. E., Fondrie, W. E., Bohaczuk, S. C., Mao, Y., Parmalee, N. L., Mallory, B. J., Harvey, W. T., Kwon, Y., Garcia, G. H., Hoekzema, K., Meyer, J. G., Cicek, M., Eichler, E. E., … Stergachis, A. B. (2024). A haplotype-resolved view of human gene regulation. bioRxiv. https://doi.org/10.1101/2024.06.14.599122
Summary of Fiber-seq inferred regulatory elements (FIREs)
For those who would prefer video I have recorded a lab meeting discussing FIRE and the methods used here. If you prefer to read please continue on.
FIREs are MTase sensitive patches (MSPs) that are inferred to be regulatory elements on single chromatin fibers. To do this we used semi-supervised machine learning to identify MSPs that are likely to be regulatory elements using the Mokapot framework and XGBoost. Every individual FIRE element is associated with an estimated precision value, which indicates the probability that the FIRE element is a true regulatory element. The estimated precision of FIREs elements are created using Mokapot and validation data not used in training. We train our model targeting FIRE elements with at least 95% precision, MSPs with less than 90% precision are considered to have average level of accessibility expected between two nucleosomes, and are referred to as linker regions.
Semi-superivized machine learning with Mokapot requires a mixed-positive training set and a clean negative training set. To create mixed positive training data we selected MSPs that overlapped DNase hypersensitive sites (DHSs) and CTCF ChIP-seq peaks. And to create a clean negative training set we selected MSPs that did not overlap DHSs or CTCF ChIP-seq peaks.
For details on running the FIRE workflow see:
For details on the outputs of the FIRE workflow see:
Running the FIRE pipeline
The FIRE pipeline is a Snakemake workflow for calling Fiber-seq Inferred Regulatory Elements (FIREs) on single molecules and peak calling with Fiber-seq.
⚠️ WARNING
This pipeline generates peak-calls and trackHubs for whole genome Fiber-seq datasets. If instead you only want to add FIRE elements to a BAM file see: ft fire. You will also want to use ft fire if your dataset is not whole genome, as the FIRE pipeline requires a whole genome dataset for the FDR calibration used in peak-calling.
Install
Please start by installing pixi which handles the environment of the FIRE workflow.
Then install FIRE using git and pixi:
git clone https://github.com/fiberseq/FIRE.git
cd FIRE
pixi install
We then recommend quickly testing your installation by running the test suite:
pixi run test
We recommend setting a Snakemake conda prefix in your bashrc, e.g. in the Stergachis lab add:
export SNAKEMAKE_CONDA_PREFIX=/mmfs1/gscratch/stergachislab/snakemake-conda-envs
Then Snakemake installs all the additional requirements as conda envs in that directory.
If you wish to distribute jobs across a cluster you may need to install the appropriate snakemake executor plugin. The SLURM executor is included in the environment (snakemake-executor-slurm)
Configuring and inputs
There are two main inputs for the FIRE workflow, a configuration file (e.g. config.yaml) and a manifest file (e.g. manifest.tbl). The configuration file contains parameters for running the workflow, a reference genome, as well as a path to the manifest file. The manifest file has two columns, the sample name (used to prefix the output files) and the path to the indexed Fiber-seq BAM (or CRAM) file for each sample.
For more details see the configuration README, the example configuration file, and the example manifest file for configuration options.
⚠️ ONT users
If you are using ONT please follow the preprocessing steps outlined here, and then be sure to include ont: true in your FIRE config file (e.g. config.yaml). This will ensure that the correct parameters are used for the ft fire command.
Run
The FIRE workflow can be executed using the pixi run fire command. Under the hood this runs a snakemake workflow and any extra parameters are passed directly to snakemake. For example:
pixi run fire --configfile config/config.yaml
If you want to do a dry run:
pixi run fire --configfile config/config.yaml -n
If you want to execute across a cluster (modify profiles/slurm-executor as needed for distributed execution):
pixi run fire --configfile config/config.yaml --profile profiles/slurm-executor
And if you want to run the FIRE workflow from another directory you can do so with:
pixi run --manifest-path /path/to/snakemake/pixi.toml fire ...
where you update /path/to/snakemake/pixi.toml to the path of the pixi.toml in the cloned FIRE repository.
You can also run snakemake directly, e.g.:
pixi shell
snakemake \
--configfile config/config.yaml \
--profile profiles/slurm-executor \
--local-cores 8 -k
Outputs of the FIRE pipeline
The FIRE pipeline returns outputs for a sample in the directory results/{sample}/ and files are labeled with the sample ({sample}) and the version of the FIRE pipeline ({v}). The following files and directories are generated:
| Output | Description |
|---|---|
| {sample}-fire-{v}-filtered.cram | CRAM file containing the all the data used in the FIRE pipeline. |
| {sample}-fire-{v}-peaks.bed.gz | BED file containing the FIRE peaks calls |
| {sample}-fire-{v}-hap-differences.bed.gz | BED file containing the results of searching for haplotype-selective peaks. |
| {sample}-fire-{v}-pileup.bed.gz | BED file containing per-base information on number of FIREs, MSPs, nucleosomes, coverage and more. |
| {sample}-fire-{v}-qc.tbl.gz | Table containing quality control metrics for the FIRE CRAM. |
| trackHub-{v}/ | Directory containing a UCSC trackHub for visualizing all the results of the FIRE pipeline. |
| additional-outputs-{v}/ | Directory containing additional outputs from the FIRE pipeline. |
More details on the individual outputs
The {sample}-fire-{v}-filtered.cram file
The CRAM file contains all the data used in the FIRE pipeline. It is a CRAM file that can be viewed with IGV or other genome browsers. Sequencing quality scores are removed from the CRAM file to reduce the file size since per base quality scores are not used in the FIRE pipeline, as well as reads with insufficient m6A signal. The CRAM file is sorted and indexed.
The {sample}-fire-{v}-peaks.bed.gz file
This is the peak file for the FIRE method. Peaks are called by identifying FIRE score (methods) local-maxima that have FDR values below a threshold. By default, the pipeline reports peaks at a 5% FDR threshold. Once a local-maxima is identified, the start and end positions of the peak are determined by the median start and end positions of the underlying FIRE elements. We also calculate and report wide peaks in the additional-outputs/ by taking the union of the FIRE peaks and all regions below the FDR threshold and then merging resulting regions that are within one nucleosome (147 bp) of one another.
The FIRE peaks file has the following columns:
| Column | Description |
|---|---|
| #chrom | Chromosome of the peak |
| peak*start | Start of the peak |
| peak_end | End of the peak |
| start | Start of the maximum of the peak |
| end | End of the maximum of the peak |
| coverage | Coverage of the peak |
| fire_coverage | Coverage of the FIREs in the peak |
| score | FIRE score of the peak (see methods) |
| nuc_coverage | Coverage of the nucleosomes in the peak |
| msp_coverage | Coverage of the MSPs in the peak |
| .**{H1,H2} | Repeats of previous columns but specific for the two haplotypes |
| FDR | False discovery rate of the peak |
| log_FDR | -10*log10 of the FDR |
| FIRE_size_mean | Mean size of the FIREs in the peak |
| FIRE_size_ssd | Standard deviation of the size of the FIREs in the peak |
| FIRE_start_ssd | Standard deviation of the start of the FIREs in the peak |
| FIRE_end_ssd | Standard deviation of the end of the FIREs in the peak |
| pass_coverage | Whether the peak passes coverage filters |
The {sample}-fire-{v}-hap-differences.bed.gz file
This file primarily contains the same columns as the FIRE peaks file but additionally has a p_value column with the results of a Fisher's exact test for the difference in coverage between the two haplotypes, and a p_adjust column with the Benjamini-Hochberg adjusted p-value. See the methods for more details.
The {sample}-fire-{v}-pileup.bed.gz file
This is a BED file containing per-base information on number of FIREs, MSPs, nucleosomes, coverage and more. The columns are calculated using ft-pileup and more details can be found in the ft-pileup documentation.
The {sample}-fire-{v}-qc.tbl.gz file
This file contains quality control metrics for the FIRE CRAM. The results are directly created by ft-qc and more details can be found in the ft-qc documentation.
The trackHub-{v}/ directory
The trackHub-{v}/ directory contains a UCSC trackHub for visualizing all the results of the FIRE pipeline. A description of the trackHub can be found in trackHub/fire-description.html. The trackHub can be loaded into UCSC by uploading the trackHub directory to a public facing website and then loading the hub.txt's URL into the UCSC trackHub browser.
A copy of the trackHub description can be found here.
The additional-outputs-{v}/ directory
The additional-outputs-{v}/ directory contains the following files:
TODO
FIRE methods
This chapter describes the methods and training used in the FIRE pipeline. There are two major components, identifying per-molecule FIRE elements, and aggregating these elements into peaks.
Training of Fire-seq inferred regulatory elements.
Training data
For training data, we generated 21 different GM12878 Fiber-seq experiments with a range of under- and over-methylated experimental conditions to ensure we captured a broad range of percent m6A (5.8-13.3%) to ensure our model could generalize to new samples with varying levels of m6A. We merged these sequencing results and randomly selected 10% of the Fiber-seq reads from 100,000 randomly selected DNase I and CTCF ChIP-seq peaks for mixed-positive labels and 100,000 equally sized regions that did not overlap DNase or CTCF Chip-seq for negative labels. We then generated features for each of these MSPs, including length, log fold enrichment of m6A, the A/T content, and windowed measures of m6A around the MSP (Supplemental Table 3) and held out 20% of the Fiber-seq reads to be used as test data.
Semi-supervised training
To carry out semi-supervised training, we used an established method, Mokapot (Fondrie & Noble, 2021; Käll et al., 2007), which we summarize below. In the first round of semi-supervised training, Mokapot identifies the feature that best discriminates between our mixed-positive and negative labels and then selects a threshold for that feature such that the mixed-positive labels can be discriminated from the negative labels with 95% estimated precision (defined below). The subset of mixed-positive labels above this threshold is then used as an initial set of positive labels in training an XGBoost model with five-fold cross-validation (Chen & Guestrin, 2016). Then this process is iteratively repeated, using the learned prediction from the previous iteration’s model to create positive labels at 95% estimated precision, until the number of positive identifications at 95% precision in the validation set ceases to increase (15 iterations, Fig. S1) (Fondrie & Noble, 2021; Käll et al., 2007).
Estimated precision of individual FIRE elements
We cannot compute the precision associated with a particular XGBoost score because we do not have access to a set of clean-positive labels. Instead, we define a notion of “estimated precision” using a balanced held-out test set of mixed-positive and negative labels (20% of the data). We defined the estimated precision (EP) of a FIRE element to be
$$ EP = 1 - \frac{FP+1}{TMP} $$
where TMP is the number of “true” identifications from the mixed positive labels with at least that element’s XGBoost score, and FP is the number of false positive identifications from negative labels with at least that element’s XGBoost score. We add a pseudo count of one to the numerator of false positive identifications so as to prevent liberal estimates for smaller collections of identifications (Fondrie & Noble, 2021).
Features of FIRE elements
The following features were used as features with Mokapot in FIRE element classification:
| Feature | Description |
|---|---|
| msp_len | Length of the MSP |
| msp_len_times_m6a_fc | Length of the MSP times the fold-change of m6A in the MSP |
| ccs_passes | Number of CCS passes in the MSP |
| fiber_m6a_count | Number of m6A sites in the Fiber-seq read |
| fiber_AT_count | Number of AT sites in the Fiber-seq read |
| fiber_m6a_frac | Fraction of m6A sites in the Fiber-seq read |
| msp_m6a | Number of m6A sites in the MSP |
| msp_AT | Number of AT sites in the MSP |
| msp_m6a_frac | Fraction of m6A sites in the MSP |
| msp_fc | Fold-change of m6A fraction in the MSP relative to the whole reads m6A fraction |
| bin_X_m6a_count | Number of m6A sites in the Xth 40 bp window |
| bin_X_AT_count | Number of AT sites in the Xth 40 bp window |
| bin_X_m6a_frac | Fraction of m6A sites in the Xth 40 bp window |
| bin_X_m6a_fc | Fold-change of m6A fraction in the Xth 40 bp window relative to the whole reads m6A fraction |
| {best, worst}_m6a_count_X | Number of m6A sites in the 100 bp window with the most (best) or least (worst) m6A sites |
| {best, worst}_AT_count_X | Number of AT sites in the 100 bp window with the most (best) or least (worst) AT sites |
| {best, worst}_m6a_frac_X | Fraction of m6A sites in the 100 bp window with the most (best) or least (worst) m6A sites |
| {best, worst}_m6a_fc_X | Fold-change of m6A fraction in the 100 bp window with the most (best) or least (worst) m6A sites relative to the whole reads m6A fraction |
There are nine 40 bp windows for each MSP (X = 1, 2, ..., 9), with the 5th window centered on the MSP.
Aggregation of FIRE elements for peak calling
Aggregate FIRE score calculation
The FIRE score (\(S_g\)) for a position in the genome (\(g\)) is calculated using the following formula:
$$ S_g=-\frac{50}{R_g} \sum_{i=1}^{C_g} \log_{10}(1-min(EP_i , 0.99)) $$
where \(C_g\) is the number of FIRE elements at the \(g\)th position, \(R_g\) is the number of Fiber-seq reads at the \(g\)th position, and \(EP_i\) is the estimated precision of the \(i\)th FIRE element at the \(g\)th position. The estimated precision of each FIRE element is thresholded at 0.99, such that the FIRE score takes on values between 0 and 100. Regions covered by less than four FIRE elements (i.e., if \(C_g < 4\)) are not scored and are given a value of negative one.
Regions of unreliable coverage
Regions with unreliable coverage are defined as regions with Fiber-seq coverage that deviate from the median coverage by five standard deviations, where a standard deviation is defined by the Poisson distribution (i.e., the square root of the mean coverage). This is a parameter that can be adjusted by the user.
FIRE score FDR calculation
We shuffle the location of an entire Fiber-seq read by selecting a random start position within the chromosome and relocating the entire read to that start position. Fiber-seq reads originating from regions with unreliable coverage are not shuffled, and reads from regions with reliable coverage are not shuffled into regions with unreliable coverage (bedtools shuffle -chrom -excl, v2.31.0). We then compute FIRE scores associated with the shuffled genome. Recalling that the FIRE score for the \(g\)th position in the genome is denoted \(S_g\), we divide the number of bases that have shuffled FIRE scores above \(S_g\) by the number of bases that have un-shuffled FIRE scores above \(S_g\). This provides an estimate of the FDR associated with the FIRE score \(S_g\).
Peak calling
Peaks are called by identifying FIRE score local-maxima that have FDR values below a 5% threshold and that exceed an optional percent actuation threshold \(C_g / R_g\). Adjacent local maxima that share 50% of the underlying FIRE elements or have 90% reciprocal overlap are merged into a single peak, using the higher of the two local maxima. Then, the start and end positions of the peak are determined by the median start and end positions of the underlying FIRE elements. We also calculated and reported wide peaks by taking the union of the FIRE peaks and all regions below the FDR threshold and then merged the resulting regions that were within one nucleosome (147 bp) of one another.
Haplotype-selective peaks
Haplotype-selective peaks
For peaks with at least 10 Fiber-seq reads on both haplotypes, we test the difference in percent actuation in each haplotype (fraction of reads with FIRE elements) using a two-sided Fisher’s exact test. Specifically, the inputs for the test are the number of FIRE elements in haplotype one, the number of Fiber-seq reads without FIRE elements in haplotype one, the number of FIRE elements in haplotype two, and the number of Fiber-seq reads without FIRE elements in haplotype two. We then apply an FDR correction (Benjamini-Hochberg) to correct for multiple testing.
Glossary of Fiber-seq terms
Fiber-seq protocol
A treatment of DNA chromatin fibers with the Hia5 enzyme which preferentially methylates accessible adenosines. The fibers are then sequenced using a long-read platform that can detect the m6A modifications.
Fiber-seq read or fiber
A DNA fiber that has been sequenced using the Fiber-seq protocol and has m6A modifications called.
m6A
N6-methyladenosine: A DNA modification made during the Fiber-seq protocol on accessible adenosines. Native m6A sites are exceedingly rare or non-existent in eukaryotic DNA.
Inferred nucleosome
A nucleosome inferred from the Fiber-seq data. The nucleosome position along the DNA is inferred by the ft add-nucleosomes algorithm using the m6A modifications (not a direct observation of a nucleosome).
MSP
Methyltransferase sensitive patch: A stretch of a Fiber-seq read that has a high density of m6A sites. Specifically it is defined as a region that is inferred to not be occluded by a nucleosome.
FIRE
Fiber-seq Inferred Regulatory Element: A MSP that is inferred to be a regulatory element based on features of the MSP including m6A density.
Internucleosomal linker region
Any MSPs that is not a FIRE element.
The Fiber-seq BAM format
The Fiber-seq BAM format adds to m6A BAM data from a PacBio (ft predict-m6a) or ONT (dorado) sequencing run with m6A.
The following tags are added to the BAM file:
ns:B:I,: Nucleosome start sites (0-based) on the forward strand of the sequencing read (u32).nl:B:I,: Equal length array tonsof nucleosome lengths (u32).as:B:I,: MSP start sites (0-based) on the forward strand of the sequencing read (u32).al:B:I,: Equal length array toasof MSP lengths (u32).aq:B:C,: Quality scores for the MSP [0, 255] (u8). This tag is optional and added byft fire.
The ns/as/nl/al tags are with respect to the unaligned sequencing read (forward strand) and are not affected by alignment or the orientation of the read after alignment. This also means that the starts and lengths encoded in these tags may change when ft lifts these molecular coordinates to reference coordinates.
The ns or as tag do not need to begin or end at the start or end of the read; however, once begun, they must be contiguous. i.e. the ns and as tags must combine to form a contiguous set of alternating nucleosome and MSP sites. The ns, nl, as, and al tags are added to the BAM file automatically using ft predict-m6a or later using the ft add-nucleosomes command.
The aq tag is added using the ft fire command and represents the estimated precision of the MSP being a FIRE. Specifically, the estimated precision of a FIRE is the value of the aq tag divided by 255.
Molecular Annotation Specification
A more comprehensive specification for molecular annotations in Fiber-seq data is being developed at fiberseq/Molecular-annotation-spec. This specification will formalize the full set of per-molecule annotations beyond the BAM tags described above.
Data Access
Broadly Consented Data Downloads
We provide access to broadly consented Fiber-seq data that can be used by researchers for various genomic studies.
Available Datasets
Our broadly consented datasets include the following samples:
- CHM1: Complete hydatidiform mole cell line
- CHM13: Complete hydatidiform mole cell line (GRCh38 alignment)
- CHM13_DSA: CHM13 data aligned against the DSA (T2Tv2.0)
- GM12878: Lymphoblastoid cell line from Coriell Cell Repository
- HG002: Human genome reference sample (Ashkenazi Jewish trio son)
- K562: Chronic myelogenous leukemia cell line
All samples are processed with FIRE (Fiber-seq Inferred Regulatory Elements).
Data Formats
All datasets are provided in standard formats:
- Fiber-seq CRAM files: Primary data format containing m6A calls and nucleosome positions
- Processed FIRE outputs: Summary statistics, peaks, and aggregated results
Download Instructions
Browse Available Data
You can explore all available samples using the AWS CLI:
aws s3 ls --no-sign-request --endpoint-url https://s3.kopah.uw.edu s3://stergachis/public/FIRE/broadly-consented/
Download Data
To download specific samples or datasets, use the AWS CLI with the following pattern:
aws s3 sync --no-sign-request --endpoint-url https://s3.kopah.uw.edu s3://stergachis/public/FIRE/broadly-consented/[SAMPLE_NAME]/ ./[LOCAL_DIRECTORY]/
For example, to download CHM13 data:
aws s3 sync --no-sign-request --endpoint-url https://s3.kopah.uw.edu s3://stergachis/public/FIRE/broadly-consented/CHM13/ ./CHM13_data/
Data Use Guidelines
When using our broadly consented data, please:
- Cite the original studies and data contributors
HPRCv2 Fiber-seq CRAMs
We also provide access to Fiber-seq CRAM for the HPRCv2 release. These CRAMs contain m6A calls, MSP calls, and nucleosome calls for each of the HPRCv2 samples with Fiber-seq data.
You can find these CRAMs in the following S3 bucket. Data is aligned both to the Donor Specific Assembly (dsa) and to GRCh38 (shared.ref).
aws s3 ls --no-sign-request --endpoint-url https://s3.kopah.uw.edu 's3://stergachis/public/HPRCv2/FIRE-bams/'
HPRCv2 FIRE Peaks
We provide pangenome graph-based union peak calls for the HPRCv2 Fiber-seq samples. These peaks are called on donor-specific assemblies and then projected to multiple coordinate systems. For full column definitions, thresholds, and methodology, see the README included in the bucket.
Available files
Union peaks (every sample × consensus peak that passes in at least one sample):
| File | Coordinates |
|---|---|
union-peaks-cons.bed.gz | Pangenome graph consensus (shown in T2T-CHM13 coords when possible) |
union-peaks-chm13.bed.gz | T2T-CHM13 reference paths |
union-peaks-asm.bed.gz | Per-sample assembly contigs |
union-peaks-hg38.bed.gz | GRCh38 liftover |
Called peaks (filtered to sites passing thresholds: FIRE coverage ≥ 4, fraction accessible ≥ 0.2):
| File | Coordinates |
|---|---|
peaks-cons.bed.gz | Pangenome graph consensus (shown in T2T-CHM13 coords when possible) |
peaks-chm13.bed.gz | T2T-CHM13 reference paths |
peaks-asm.bed.gz | Per-sample assembly contigs |
peaks-hg38.bed.gz | GRCh38 liftover |
All files are bgzipped BED files with a #chrom header line and tabix indices (.tbi).
Download
Browse available files:
aws s3 ls --no-sign-request --endpoint-url https://s3.kopah.uw.edu 's3://stergachis/public/HPRCv2/FIRE-peaks/'
Download all peak files:
aws s3 sync --no-sign-request --endpoint-url https://s3.kopah.uw.edu s3://stergachis/public/HPRCv2/FIRE-peaks/ ./FIRE-peaks/
Cite
If you use fibertools in your research, please cite the following paper:
Jha, A., Bohaczuk, S. C., Mao, Y., Ranchalis, J., Mallory, B. J., Min, A. T., Hamm, M. O., Swanson, E., Dubocanin, D., Finkbeiner, C., Li, T., Whittington, D., Noble, W. S., Stergachis, A. B., & Vollger, M. R. (2024). DNA-m6A calling and integrated long-read epigenetic and genetic analysis with fibertools. Genome Research. https://doi.org/10.1101/gr.279095.124
If you use FIRE in your research, please cite the following paper:
Vollger, M. R., Swanson, E. G., Neph, S. J., Ranchalis, J., Munson, K. M., Ho, C.-H., Sedeño-Cortés, A. E., Fondrie, W. E., Bohaczuk, S. C., Mao, Y., Parmalee, N. L., Mallory, B. J., Harvey, W. T., Kwon, Y., Garcia, G. H., Hoekzema, K., Meyer, J. G., Cicek, M., Eichler, E. E., … Stergachis, A. B. (2024). A haplotype-resolved view of human gene regulation. bioRxiv. https://doi.org/10.1101/2024.06.14.599122
Analyses of Fiber-seq data
This notebook contains some of the prepublication analyses of Fiber-seq data we have done:
ONT Fiber-seq Analysis
Matched ONT and PacBio preparations
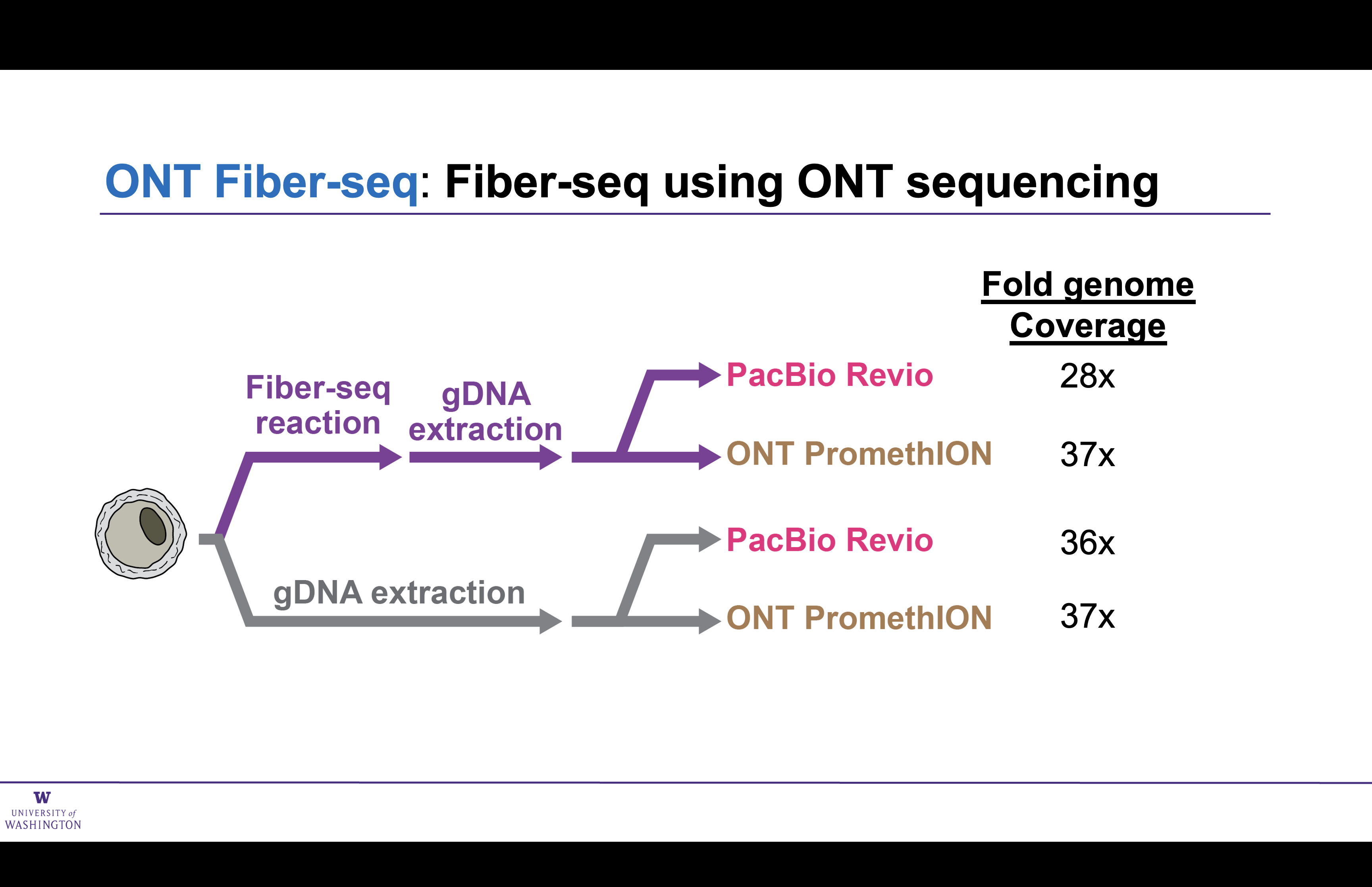
Read lengths unchanged by Fiber-seq
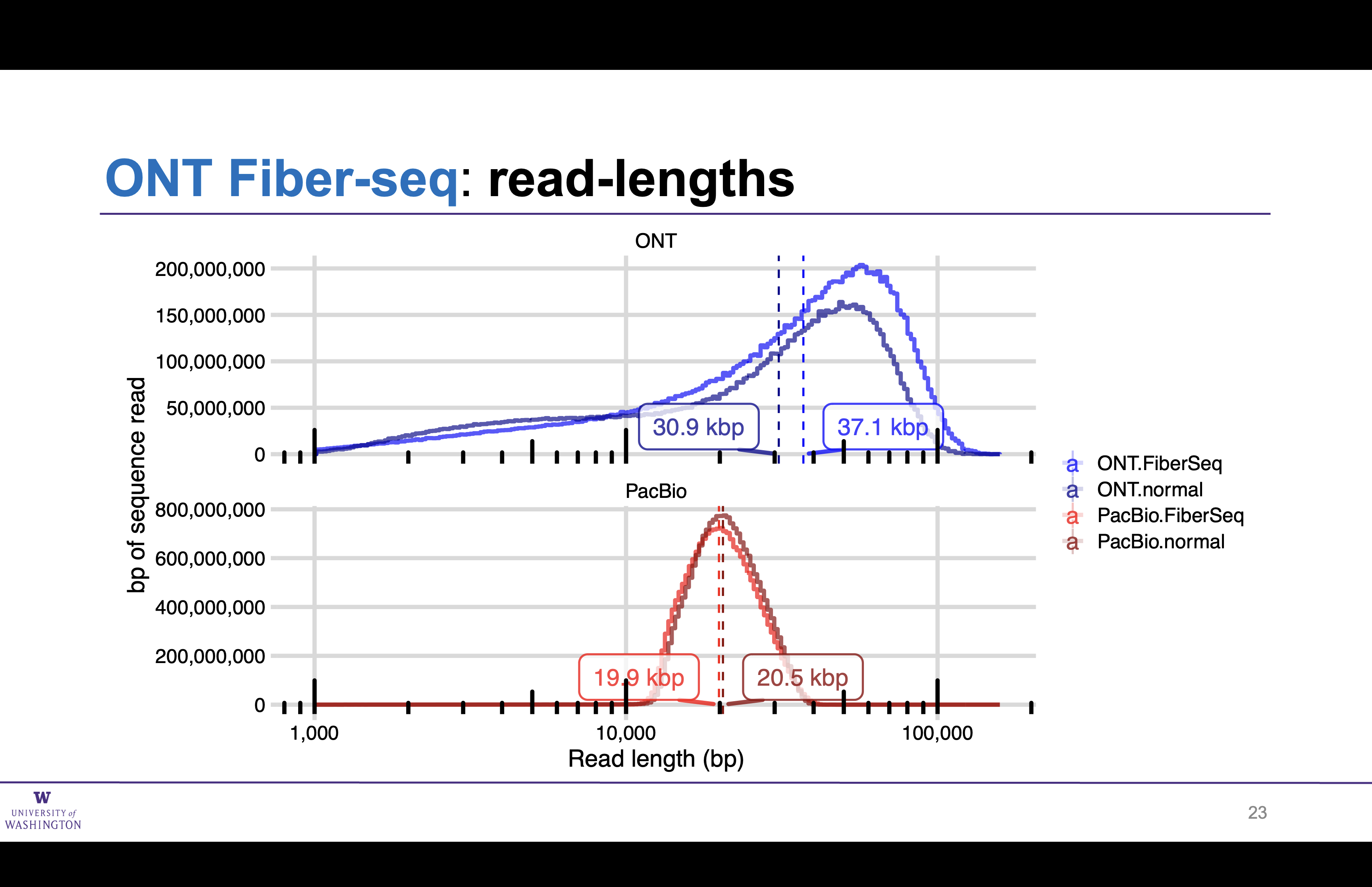
Impact of Fiber-seq on ONT read quality
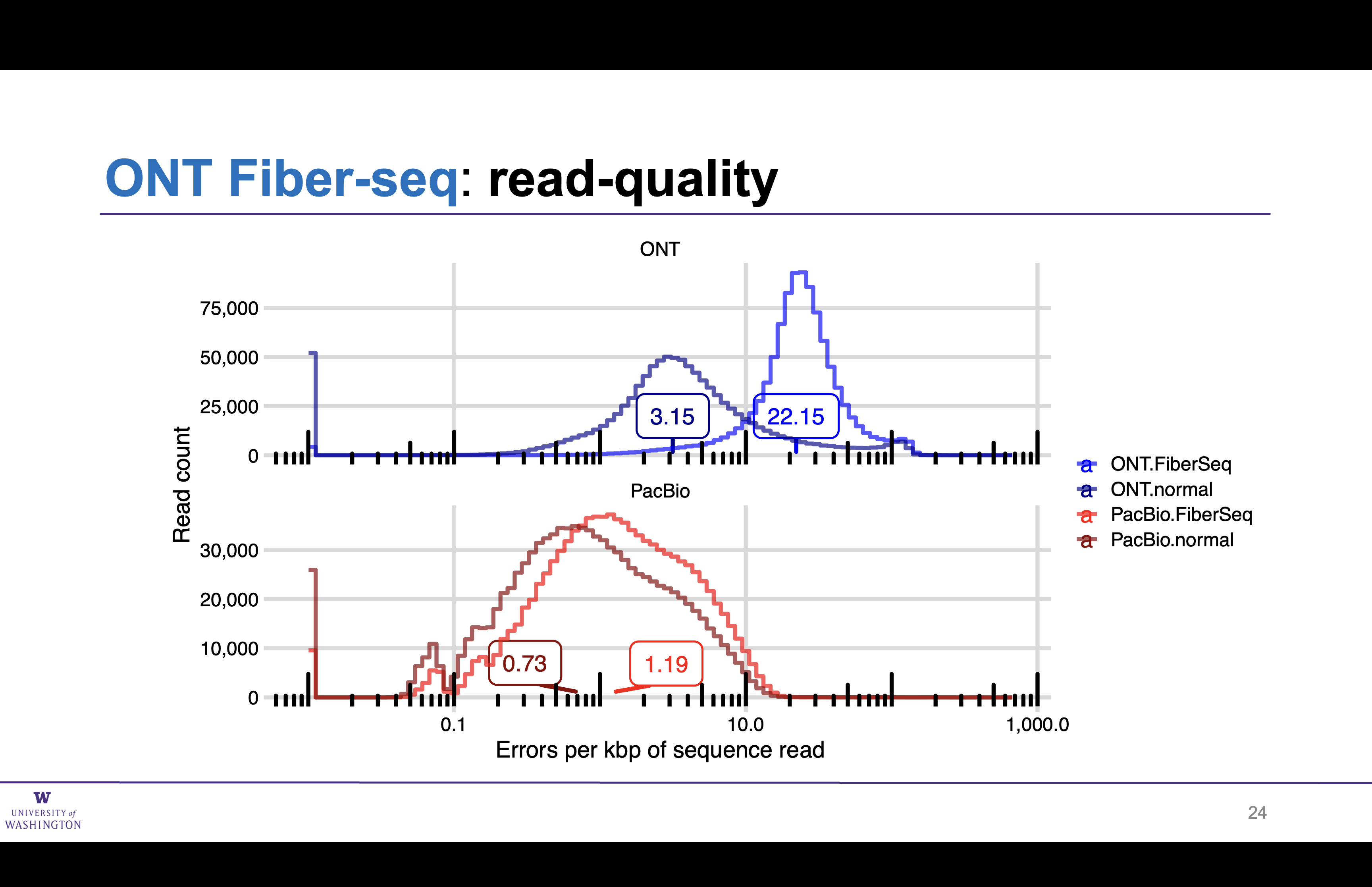
A comparison of ONT and PacBio Fiber-seq
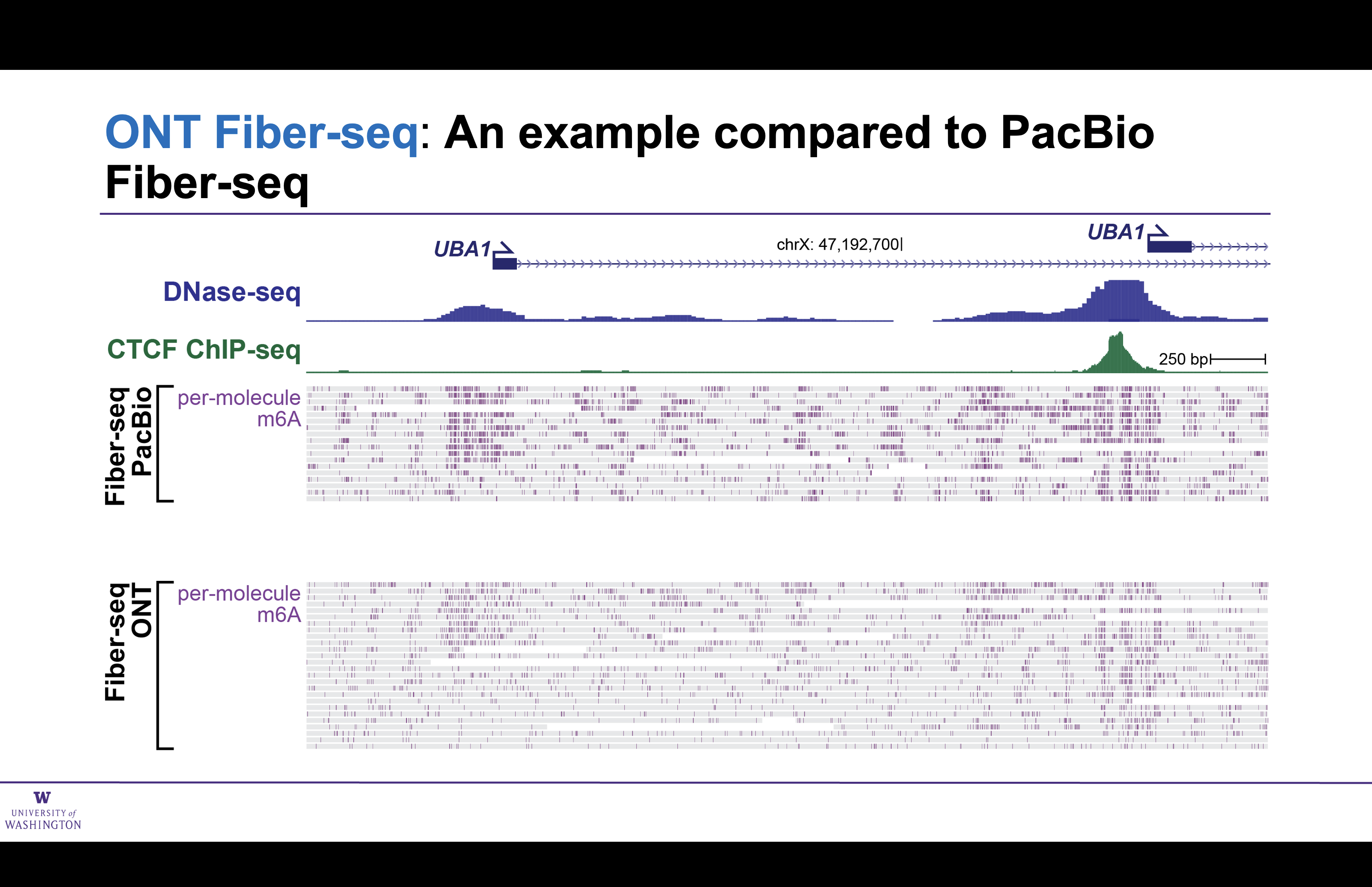
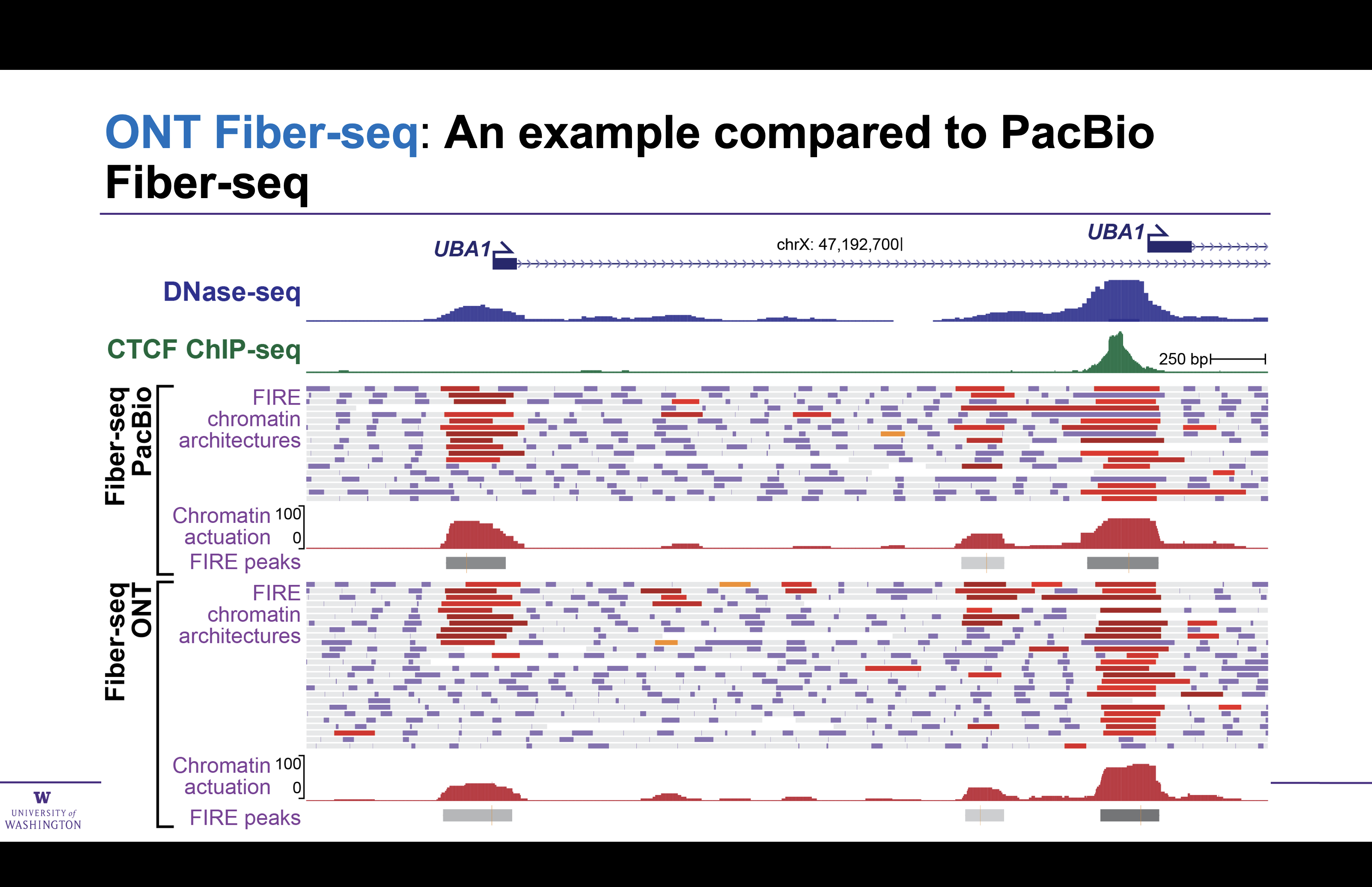
Genome-wide comparison of ONT and PacBio Fiber-seq
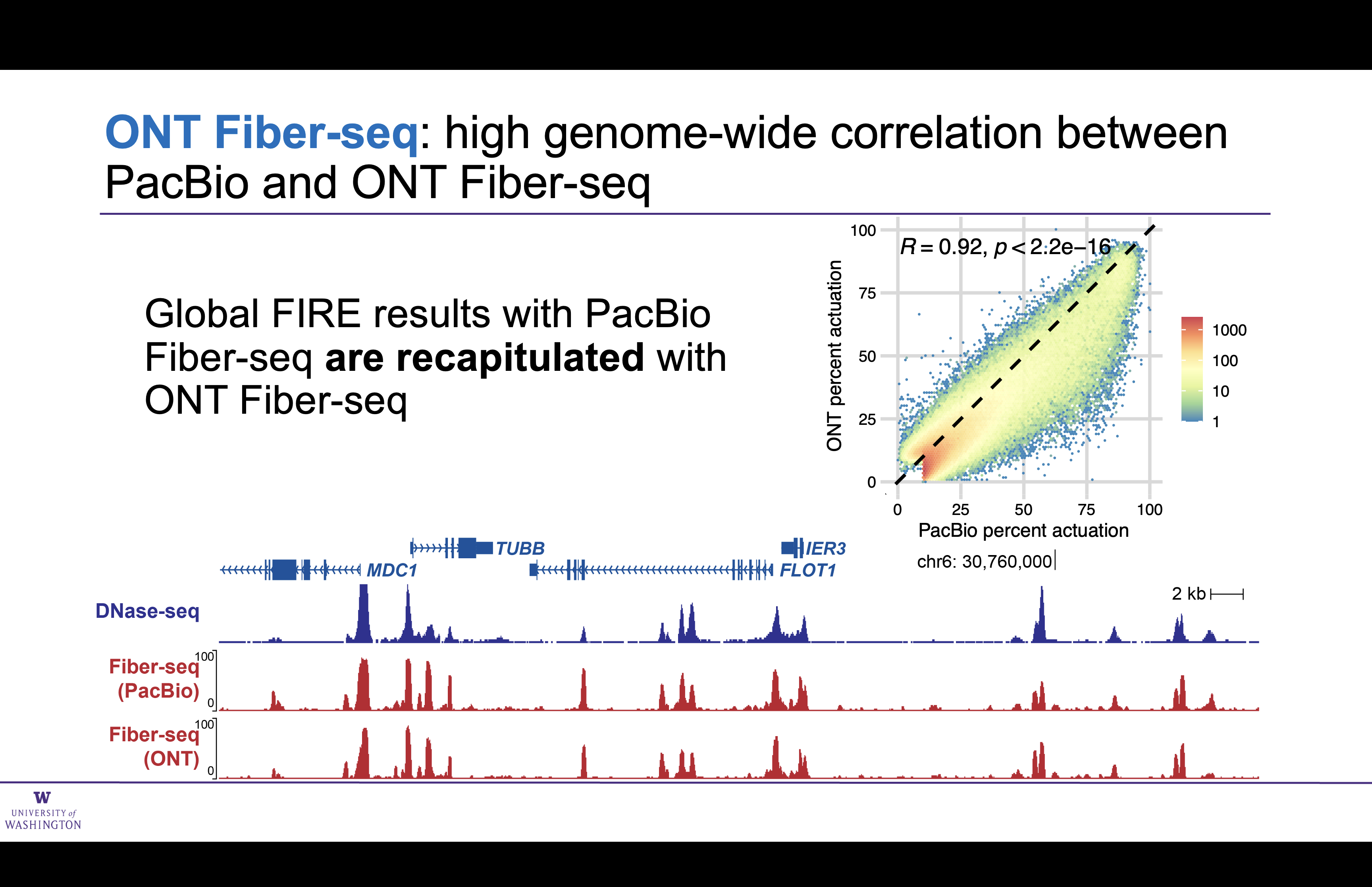
Transcription factor foot printing has reduced resolution with ONT
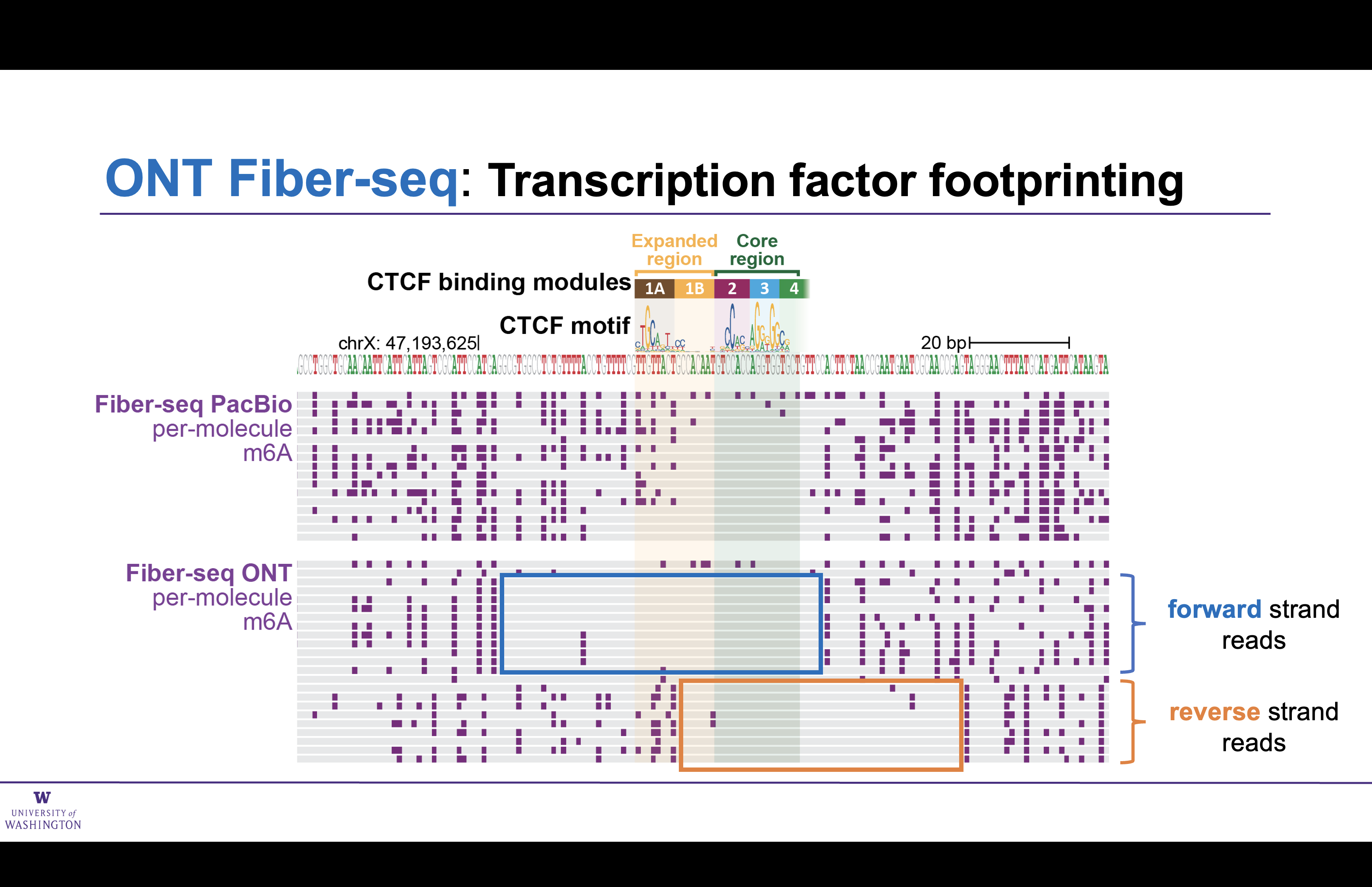
Stand does not significantly impact ONT Fiber-seq FIRE results
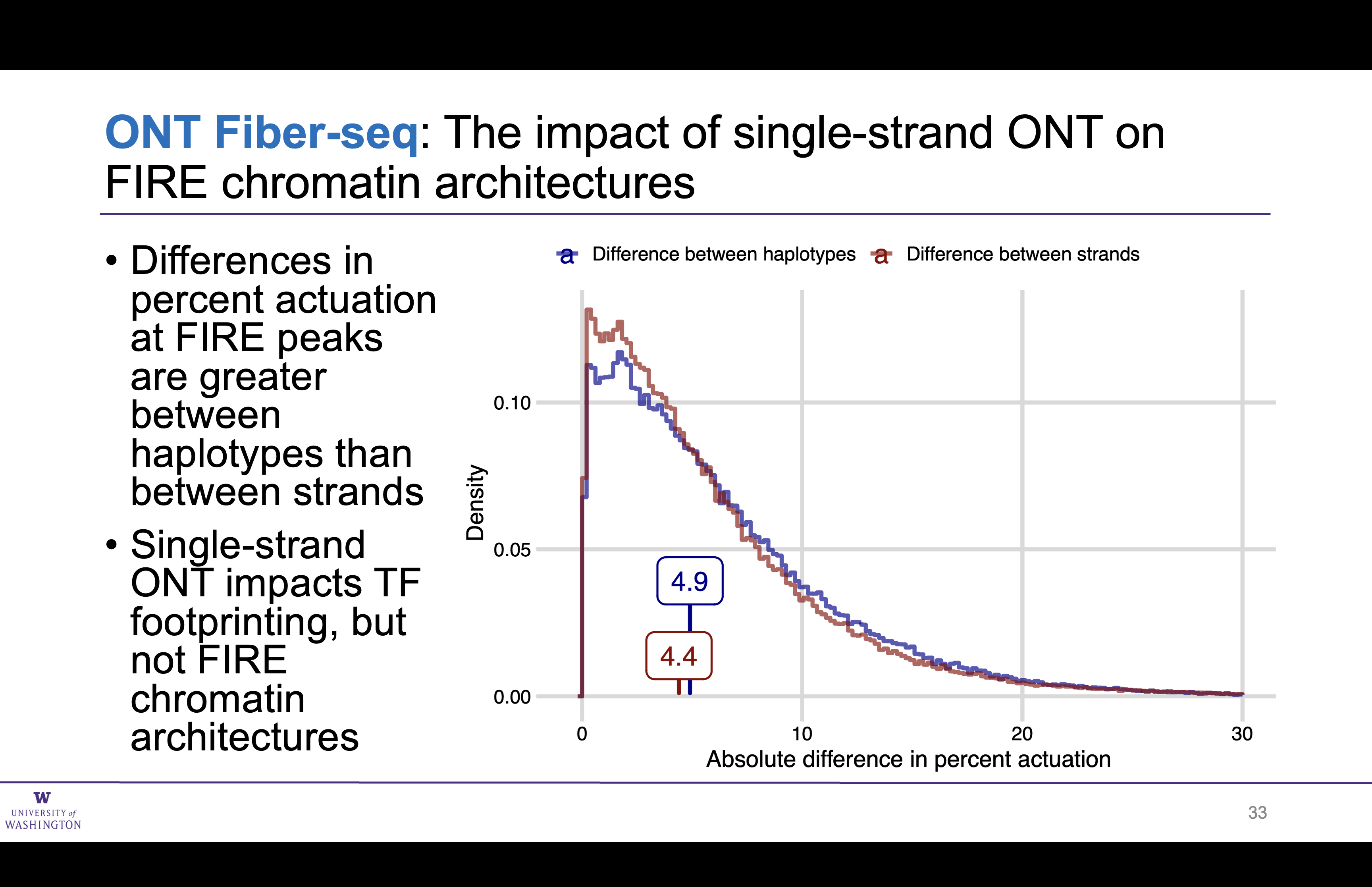
ONT FIREs have ~half as many m6A sites as PacBio (as expected)
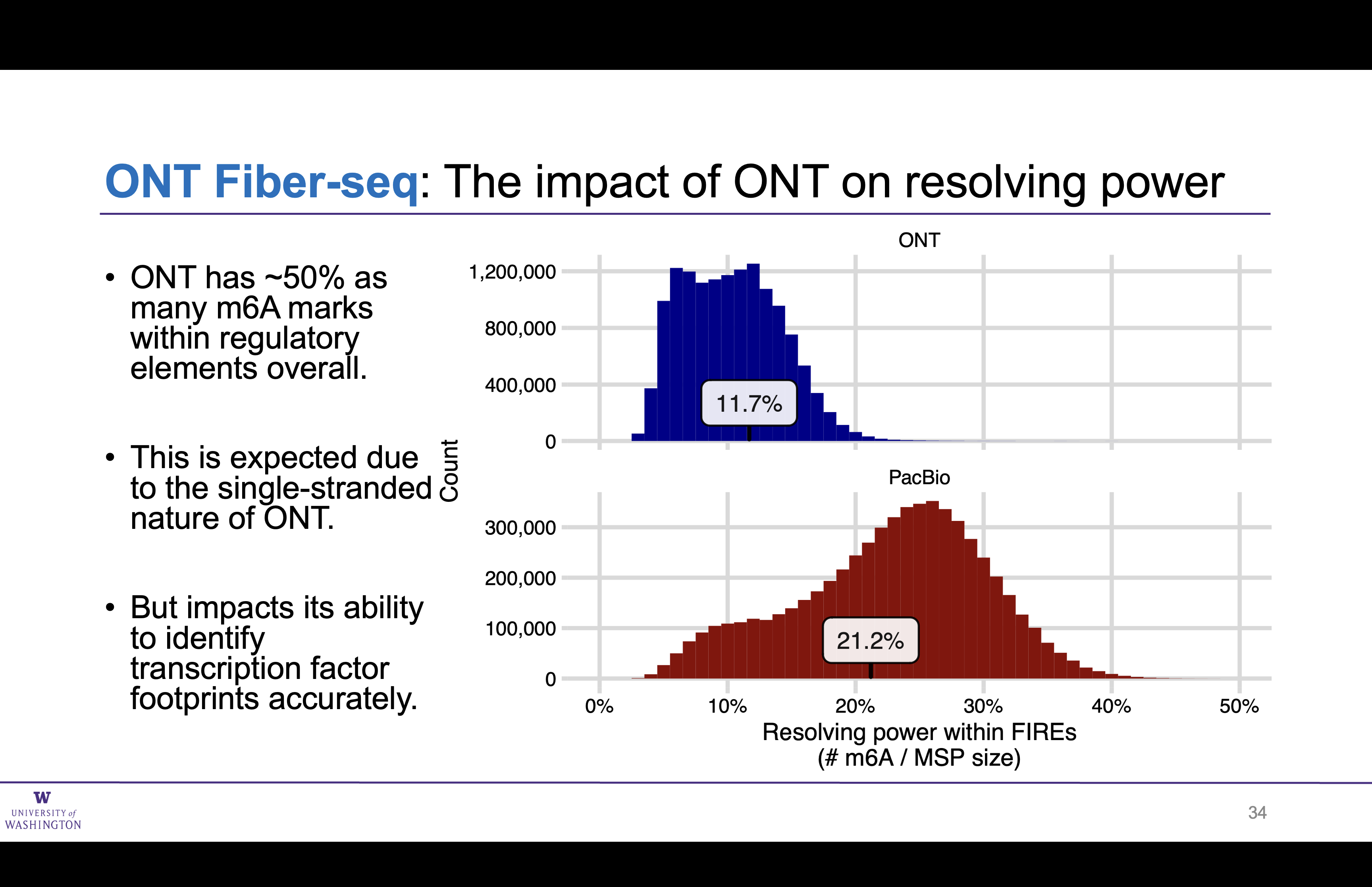
Auto-correlation of m6A sites in ONT nearly matches a single strand of PacBio
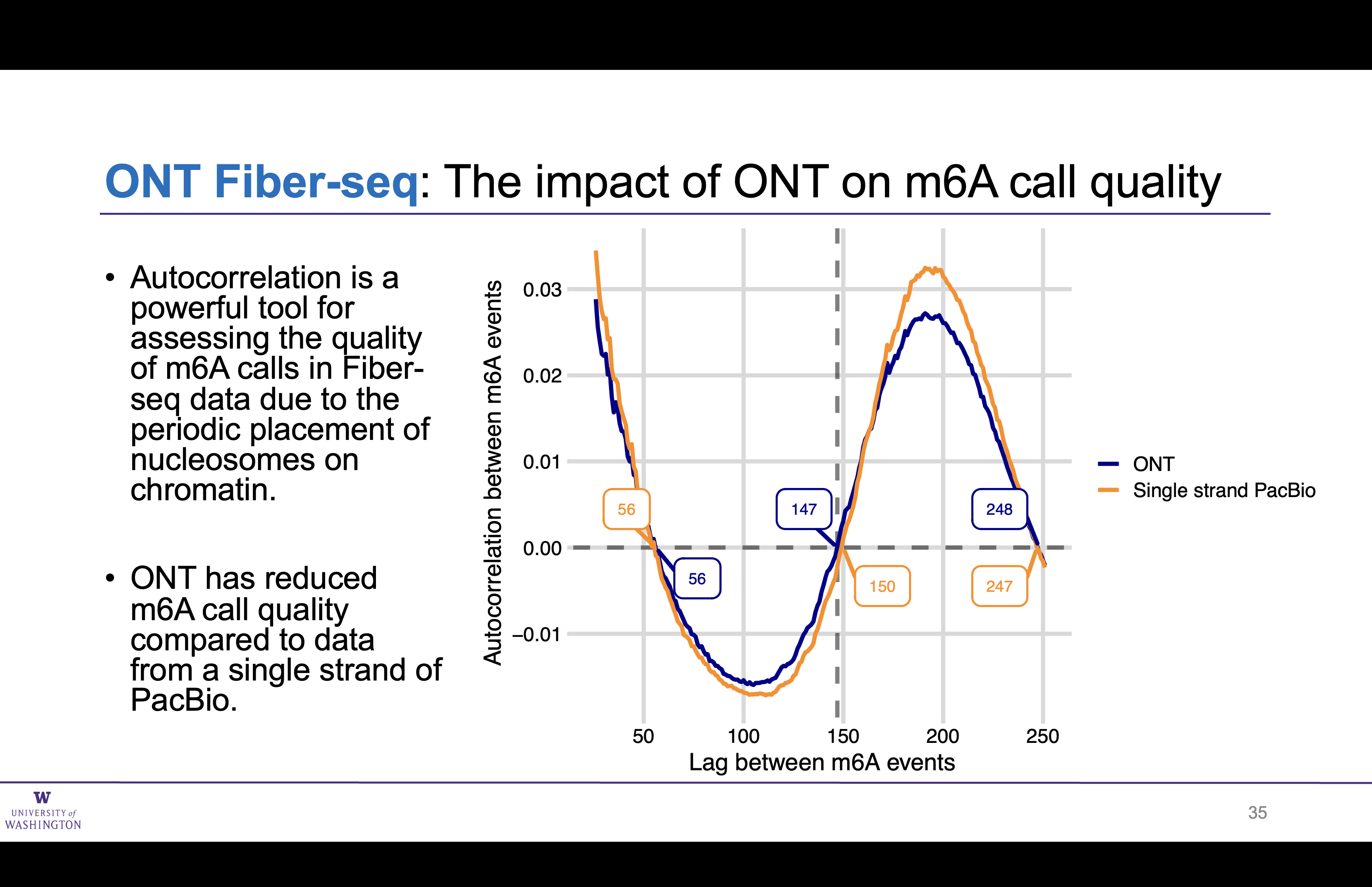
Overall conclusions
