The Guide to DAF-seq
DAF-seq (Deaminase-Assisted single-molecule chromatin Fiber sequencing) is a method that enables single-molecule footprinting at near-nucleotide resolution while synchronously profiling single-molecule chromatin states and DNA sequence. It leverages SsDddA, a nonspecific cytidine deaminase from Simiaoa sunii, to selectively stencil single-molecule protein occupancy on intact nuclei. DAF-seq is compatible with both PacBio HiFi and Oxford Nanopore sequencing platforms, and a single-cell version (scDAF-seq) is also available.
The method is described in:
Swanson, Mao, Mallory, Vollger, Bohaczuk, Oliveira, Lyon, Ranchalis, Parmalee, Cohen, Bennett & Stergachis. Mapping single-cell diploid chromatin fiber architectures using DAF-seq. Nature Biotechnology (2025). DOI: 10.1038/s41587-025-02914-3
Quick start
- Running the experiment? Start with the DAF-seq Protocol for the complete wet lab workflow.
- Analyzing data? Jump to the DAF-QC Pipeline for computational setup and quality control.
- Calling footprints? Run FiberHMM to call nucleosomes, MSPs, and TF/Pol II footprints from DAF-seq or Fiber-seq data.
- Exploring your data? Use FiberBrowser to visualize and analyze DAF-seq and Fiber-seq reads interactively.
- New to DAF-seq? See the Glossary for definitions of key terms.
What's here
Wet Lab
- DAF-seq Protocol -- Complete protocol for nuclei isolation, on-nuclei cytosine deamination, and DNA extraction
- Primer Design -- Guidelines for designing primers for targeted DAF-seq
- Protein Purification -- Purification protocols for SsDddA and DddI
Dry Lab
- DAF-QC Pipeline -- Snakemake pipeline for quality control and initial processing of DAF-seq data
- FiberHMM -- HMM caller for nucleosomes, MSPs, and TF/Pol II footprints from DAF-seq and Fiber-seq data
- FiberBrowser -- Interactive single-molecule genome browser for DAF-seq and Fiber-seq data
Reference
- Glossary -- Definitions of key DAF-seq terms and concepts
- Cite -- Citation information and BibTeX entry
Contributing
You can help improve this guide! Click the edit button () in the top right of any page to suggest changes.
For questions or contributions, see the relevant GitHub repositories:
- DAF-seq Manuscript -- Analysis code and supplementary materials
- DAF-QC-SMK -- QC pipeline source code and documentation
- This website -- Source for these documentation pages
DAF-seq Protocol
This protocol covers the complete DAF-seq workflow from nuclei isolation through DNA extraction. For primer design guidelines, see Primer Design. For SsDddA and DddI purification, see Protein Purification. After library preparation and sequencing, use the DAF-QC Pipeline for data processing and quality control.
Reagents & Buffers
Buffer A
| Final Concentration | Stock Concentration | Volume stock soln |
|---|---|---|
| RNase- DNase- free H2O | 100% | 960 uL |
| 15 mM Tris-Cl, pH 8.0 | 1 M Tris-Cl, pH 8.0 | 15 uL |
| 15 mM NaCl | 5 M NaCl | 3 uL |
| 60 mM KCl | 3 M KCl | 20 uL |
| 1 mM EDTA, pH 8.0 | 0.5 M EDTA, pH 8.0 | 2 uL |
| 0.5 mM EGTA, pH 8.0 | 0.5 M EGTA, pH 8.0 | 1 uL |
| 0.5 mM Spermidine | 0.5 M Spermidine | 1 uL |
Note: Buffer A without spermidine can be prepared in large quantities (e.g. 100 mL) and safely stored for up to 6 months at room temperature. Add 1 uL of 0.5 M spermidine to 999 uL of buffer A stock right before using. Store spermidine at -20 C for up to 6 months.
Lysis buffer
To be optimized by user. Below are examples that have been used successfully in our lab
Cell lines: 2X Lysis buffer
| Cell Line | Final % IGEPAL | 2X Stock solution | µl 10% IGEPAL/1ml Buffer A |
|---|---|---|---|
| K562 cells | 0.025 | 0.05 | 5 µl |
| HepG2 | 0.025 | 0.05 | 5 µl |
| Lymphoblasts | 0.025 | 0.05 | 5 µl |
| HeLa | 0.05 | 0.1 | 10 µl |
| Fibroblasts | 0.1 | 0.2 | 20 µl |
Tissue: Homogenization buffer
| Final Concentration | Stock Concentration | Volume stock soln |
|---|---|---|
| RNase- DNase- free H2O | 100% | 716 µL |
| 250mM Sucrose | 1.5M Sucrose | 167 µL |
| 15 mM Tris-Cl, pH 8.0 | 1 M Tris-Cl, pH 8.0 | 15 µL |
| 15 mM NaCl | 5 M NaCl | 3 µL |
| 60 mM KCl | 2 M KCl | 30 µL |
| 1 mM EDTA, pH 8.0 | 0.5 M EDTA, pH 8.0 | 2 µL |
| 0.5 mM EGTA, pH 8.0 | 0.5 M EGTA, pH 8.0 | 1 µL |
| 0.5 mM Spermidine | 0.5 M Spermidine | 1 µL |
| 0.1 mM DTT | 100mM DTT | 10 µL |
| 0.1% Triton X-100 | 10% Triton X-100 | 30 µL |
| 1x Protease inhibitor | 50x in EtOH (Promega G6521) | 20 µL |
| 0.2U RNasein plus | 40U/ul RNasein plus | 5 µL |
Note: Nuclei isolation is cell and tissue-type dependent and should be optimized by the user to ensure completion. Digitonin is also compatible.
Additional reagents
- 1x PBS, pH 7.4 + 0.5% BSA (for cells)
- UNG inhibitor (NEB M0281)
- SsDddA stock*: 100 uM, aliquots stored at -80 C
- DddI stock*: 1000 uM, aliquots stored at -80 C
- NEB Monarch Spin gDNA Extraction Kit (T3010)
- Qubit dsDNA assay
- LoBind tubes
* See Protein Purification for how to generate these. You can also e-mail absterga@uw.edu for protein aliquots
Nuclei Isolation
Example for cells
Below is an example protocol which we have used for LCLs. Nuclei isolation should be optimized by the user for each tissue and cell type.
- Collect 250k-1M cells into a 1.5 mL LoBind tube.
- Pellet at 350 x g for 5 min at 4 C. Remove supernatant.
- Wash cells with 1 mL 1x PBS, pH 7.4 + 0.5% BSA.
- Pellet at 350 x g for 5 min at 4 C. Remove supernatant.
- Resuspend pellet in 60 uL Buffer A.
- Add 60 uL 2x Lysis Buffer (1:1 dilution to a final 1× Lysis Buffer concentration). Mix by gently tapping the side of the tube and incubate 10 min on ice.
- Spin 350 x g for 5 min at 4 C. Remove supernatant.
- Resuspend nuclei in 50 uL Buffer A.
IMPORTANT: Ensure that cells are fully lysed before proceeding to the next step.
- Count 100k-250k nuclei and add Buffer A + 1 uL UNG inhibitor (2 U) to a final volume of 47 uL.
- Continue to On-Nuclei Cytidine Deamination
Example for tissues
Below is an example protocol which we have used for tissues including liver and brain. Nuclei isolation should be optimized by the user for each tissue and cell type. Some tissues such as muscle may require pulverization with LN2 in a mortar prior to douncing
- Add 1mL homogenization buffer and tissue to dounce, keep on ice.
- Homogenize the tissue with:
- 10 strokes of the loose pestle A (avoid adding too much pressure and avoid foaming)
- 10 strokes of the tight pestle B (avoid adding too much pressure and avoid foaming)
- Using a wide bore P1000 pipette tip, transfer homogenate out of the dounce and pass it through a 70µm cell strainer into a 15 ml low bind tube.
- Centrifuge 350xg for 5 min at 4°C.
- Discard supernatant. Keep pellet.
- Resuspend nuclei in 50 µL cold buffer A. Mix gently and count nuclei.
IMPORTANT: Ensure that cells are fully lysed before proceeding to the next step
- Count 100k-250k nuclei and add Buffer A + 1 µL UNG inhibitor (2 U) to a final volume of 47 µL.
- Continue to On-Nuclei Cytidine Deamination
On-Nuclei Cytidine Deamination
-
Add 1-2 µL SsDddA (100 µM stock, final 2-4 µM) to the 47 µL nuclei suspension.
-
Incubate 10 min at 25°C in a PCR machine or heat block. Mix gently; avoid vortexing.
-
Immediately add 1 µL DddI (1000 µM, 5-molar excess) to stop the reaction. Mix by gently tapping.
-
Proceed according to your application:
a. Targeted DAF-seq: Proceed to DNA Extraction for Targeted DAF-seq. Deaminase reactions may be safely stored at -20°C.
b. Single-cell DAF-seq: Proceed to Single-cell DAF-seq Fluorescence-Activated Cell Sorting (FACS). Maintain nuclei on ice until sorting.
IMPORTANT: Enzyme activity varies by batch. We are currently in the process of defining an activity unit. For our current batch of SsDddA (as of April 2026, produced by our lab in mid-2025), we recommend starting with 2 uM and further optimizing concentration by enzyme titration, if needed.
DNA Extraction for Targeted DAF-seq
Follow the manufacturer's instructions. The steps below reflect the lab's working sequence for samples after the on-nuclei deamination stop. For routine targeted DAF-seq applications, we recommend NEB Monarch Spin gDNA Extraction Kit (T3010) as the length of isolated DNA is typically sufficient for the amplification of 2-7 kb products. A HMW kit may be necessary for longer range PCR products.
- Add PBS to bring the total volume to 100 uL (typically 50 uL).
- Add 1 uL Proteinase K and 3 uL RNase A; mix by brief vortexing.
- Add 100 uL Cell Lysis Buffer; vortex 10 s.
- Incubate 5 min at 56 C in a thermal mixer with agitation (~1400 rpm).
- Add 400 uL gDNA Binding Buffer; vortex 5-10 s.
- Centrifuge briefly (~1 min at 16,000 x g) if needed to collect contents; transfer to a gDNA purification column seated in a collection tube.
- Close cap and centrifuge 3 min at 1,000 x g to load/bind DNA onto the column.
- Immediately centrifuge 1 min at 16,000 x g to clear the membrane.
- Discard collection tube and place column into a fresh collection tube.
- Add 500 uL gDNA Wash Buffer; centrifuge 1 min at >=12,000 x g.
- Discard flow-through and repeat the 500 uL Wash Buffer step and spin.
- Transfer column to a clean 1.5 mL LoBind tube.
- Add 35 uL pre-warmed (56 C) gDNA Elution Buffer directly to the membrane.
- Let stand 1 min at room temperature; centrifuge 1 min at >=12,000 x g to elute DNA.
- Store eluted gDNA at 4 C (short-term) or -20 C (long-term). Quantify with Qubit.
The DNA is now ready for PCR.
Single-cell DAF-seq Fluorescence-Activated Cell Sorting (FACS)
- Aliquot 3 ul of BioSkryb Cell Buffer into desired wells of a 96-well PCR plate. Leave first or last column empty to allow space for BioSkryb PTA controls.
- Create forward scatter (FSC-A & FSC-H) and side scatter (SSC-A) gates to remove doublets and debris.
- Sort individual nuclei into plate wells containing BioSkryb Cell Buffer.
- Seal and quickly centrifuge the nuclei plate.
- Flash freeze the nuclei plate at -80C or dry ice and store until PTA amplification.
Notes
- The reaction volume of the "On-Nuclei Cytidine Deamination" may be doubled to accommodate 500K nuclei in step 9. If 100 uL volume is used, no additional buffer is required at step 14 of "DNA Extraction".
- We have observed that the recovery of SsDddA-treated DNA is approximately 10-20% as efficient as untreated DNA using the Monarch Spin gDNA Extraction Kit. This recovery is typically sufficient for downstream applications.
- Buffer A is compatible with SsDddA provided by our lab. Other cytidine deaminases may require buffers containing ZnCl2.
Next steps
After library preparation and sequencing, process your data with the DAF-QC Pipeline to assess targeting efficiency, deamination rates, strand calling, and other quality metrics.
Primer Design
A strategy for designing primers for targeted DAF-seq experiments.
FiberBrowser also provides a built-in primer design tool (local BLAST required) that can identify the best DAF-compliant primers covering a selected region, letting you use your existing Fiber-seq data as a guide.
Materials
- Untreated gDNA: We recommend a median size of ~20 kb for most applications, which we routinely isolate using the Monarch Spin gDNA Extraction Kit (T3010).
- DAF-seq DNA: SsDddA-treated chromatin followed by DNA extraction (see DAF-seq Protocol). For primer validation, chromatin from a readily available/cheap source that contains the target locus of interest should be used (e.g. cell lines), although the target sample is preferred when possible.
- Uracil-tolerant DNA polymerase/PCR master mix: It is critical to use a DNA polymerase that tolerates uracil in the template. We have had the best results with RepliQa HiFi ToughMix (QuantaBio, 95200). Other uracil-tolerant polymerases we have tested with varying results include Q5U (NEB M0597), PhusionU (Thermo Scientific F562), LongAmp (NEB M0323), and KOD One (Toyobo KMM-101).
- PCR cleanup kit: We use Monarch Spin PCR & DNA Cleanup Kit (NEB T1130), but most kits should work as long as they can process the proper PCR fragment size.
Identify a target window
Targeted DAF-seq is typically performed to amplify 2-10 kb targets using PCR (see Choosing a product length below). We recommend that primer binding sites are designed to be at least 100 bp outside of the genomic element of interest (preferably >500 bp). Below are features to consider when designing primers (in order of priority):
- When possible, keep multiple regulatory elements within the same amplicon if co-actuation is to be considered as part of the analysis.
- Avoid placing primer binding sites directly within accessible chromatin regulatory elements within your sample of interest, which can typically be determined using existing orthogonal datasets such as ATAC-seq, DNase-seq, or Fiber-seq.
- Avoid placing primer binding sites within repetitive elements, which can be identified using the RepeatMasker track in the UCSC genome browser.
- Avoid placing primer binding sites directly over common germline genetic variants within your species of interest. If you are using human samples for which a germline VCF is not available, avoid common variants, which can be displayed in the UCSC browser gnomAD track (be sure to select "All" annotation types; we recommend a threshold of 0.01).
- Consider avoiding primer binding sites that result in internal repetitive elements with multiple copies within the amplified region. In some cases, homologous repeats can cause deletions within PCR products. This is easily detected by shallow sequencing and should not be prioritized in the first round of design if it compromises inclusion of desired elements.
Design candidate primers
Primer design for DAF-seq requires unique considerations due to template deamination. The guidelines below represent a primer design strategy using NCBI primer-BLAST followed by curation.
As each region and deamination pattern is unique, it may not always be possible to satisfy all recommendations:
-
Set design parameters: Paste the target region and specify the positions in which to design primers. Set the PCR product size to the desired range. You may also broaden the "Primer melting temperatures" if few primers are returned.
In the "Primer Pair Specificity Checking Parameters", change "Database" to "Refseq representative genomes", and increase the "Max target amplicon size" as appropriate (typically a few kb higher than the intended target).
In "Advanced parameters", change "Primer Size" to 20-28 (optimal 23). Change "Primer GC content(%)" to 20-50%. Change "Max Poly-X" to 3 (see Poly-X stretches). Change "Max GC in primer 3' end" to 3.
-
Curate primers for the following:
- Specificity (critical): For primers with identified off-target sites, pay special attention to those where a C>T mutation would make a better primer binding site. Reject primer candidates with many partial off-target matches.
- C/G in 3' end (critical): Prefer primers with the absence of C/G in the terminal 3 bases on the 3' end, although primers with the absence of C/G in the terminal 2 bases have also been successful for DAF-seq.
- Minimize C/G content (preferred): When possible, prefer primers with <40% C/G.
- Balance C/G (optional): See C and G in primers.
-
Select primer sets for testing: From our experience, less than 50% of selected primer sets will successfully amplify a single band with SsDddA-treated template. For this reason, we recommend selecting several primer sets per target to accelerate screening.
PCR annealing temperature gradient with gDNA
Critical: Ensure that the DNA polymerase tolerates uracil in the template. We have had the best results with RepliQa HiFi ToughMix (QuantaBio, 95200). Other uracil-tolerant polymerases include Q5U, PhusionU, LongAmp, and KOD One.
Perform an annealing temperature gradient on primer candidates using untreated gDNA template. We prefer to use gDNA for validation at this point both because gDNA is cheaper and easier to produce and because primer binding sites are intentionally designed outside of known regulatory elements, so the majority of the final DAF-seq template should be nucleosome-protected and unmodified, as is simulated by gDNA. Basically, if it doesn't work on gDNA, it won't work for DAF-seq.
Look for primer sets that produce a single band of the anticipated size in agarose gel electrophoresis. Low off-target amplification may be tolerable if primer choices are limited. If multiple temperatures produce comparable amplification of the single product, we prefer the lowest temperature which should theoretically have more tolerance for deaminations in DAF-seq DNA.
If multiple primer sets are successful at this stage, we recommend comparing the performance of all of them through sequencing on DAF-seq template.
Validate PCR with DAF-seq template and shallow sequencing
Perform PCR amplification on DAF-seq template with the best primer sets and annealing temperatures determined in the previous step. We recommend a 50 uL reaction. Run ~10 uL on an agarose gel to validate the presence of a single band. Off-target amplification may be tolerable if primer choices are limited.
Purify the remaining product with a PCR cleanup kit. We do not recommend gel extraction as the buffers can interfere with subsequent sequencing.
Perform shallow sequencing with a long-read method. We typically use the ONT-based Plasmidsaurus Premium PCR (standard) for routine validation. In our experience, primers that perform well in shallow ONT-based sequencing have also performed well on full-scale PacBio sequencing.
Run QC to evaluate primer performance
Run the DAF-QC-SMK pipeline to evaluate primer performance. Optimal primers will have:
- Low (<10%) off-target amplification
- A high proportion of full-length reads (>80%)
- Approximately 50/50 ratio of CT/GA reads, although primer sets with substantial strand bias may be acceptable for some applications (such as when identifying single nucleotide variants is not critical)
Also be sure to visualize reads in IGV. Check for any internal deletions and evaluate whether they are haplotype-specific. Non-haplotype-specific deletions may represent PCR artifacts that can be resolved by tuning PCR conditions or redesigning primer binding sites to exclude problematic repetitive regions (see point 5 in Identify a target window).
Additional considerations
Primer design rules are not rigid
Primer design for DAF-seq can be much more difficult than typical PCR primer design. Identifying primer sets with high specificity is challenged by the presence of template modifications and the preference to minimize C/G content. For the most difficult targets, a flexible approach that bypasses some of the recommendations above may be necessary.
Nucleosome protection is an asset
When a regulatory landscape is known through alternative datasets, it is important to avoid primer binding sites overlapping accessible elements. With this in mind, most of the template DNA will be nucleosome-occluded as nucleosome footprints are ~147 bp, while internucleosomal linker regions (i.e. accessible regions between nucleosomes that are not regulatory elements) are closer to ~10 bp. This means that most template strands will be complementary to primers even within C/G positions.
Degenerate bases in primers
To account for the possibility that there could be deaminations within primer binding sites, our initial publication used primers designed with degeneracy at G genomic positions in the form of randomly incorporated G or A (IDT). Given the point above regarding nucleosome protection, we later reconsidered this design and tested whether degenerate bases facilitate amplification. In general, we found that targets amplified better without degenerate bases.
Selection of PCR polymerase
It is critical to select a polymerase tolerant of uracil in the DNA template. This is one of the most common mistakes. Polymerase fidelity will also impact the quality of results, especially if deduplication is desired as spurious mutations can cause duplicates to be missed. The polymerase must also be suitable for relatively long-range amplifications, although this is quite common. We have found that RepliQa HiFi ToughMix (QuantaBio, 95200) fulfills these requirements and has been relatively robust in our hands.
Locked nucleic acids (LNAs) for difficult targets
In specialized cases, we have used primer sets containing locked nucleic acids to boost product yield and annealing temperature (e.g. when needed for compatibility in multiplexed assays or for C/G-rich templates prone to forming secondary structures). We have found that primers containing a single LNA are tolerated in PacBio and ONT sequencing, but adding more than one LNA causes substantial sequencing dropout with PacBio (ONT not tested). As LNA-containing primers are substantially more expensive, we recommend adding them only when a faint on-target band is already observed and multiple primer sets have failed.
In our hands, we have observed improved yield with LNAs positioned within 5 bases of the 3' and 5' ends, and we typically add them between A/A, A/T, or T/T bases. The effects of LNAs on primer specificity are complex, and LNA-containing primers have only been superficially explored in our lab. We recommend further literature review for users requiring LNA-containing primers.
Multiplexed assays
We have successfully designed primer sets for multiplexed PCR, although this can be extremely challenging. Try to design primers with comparable predicted annealing temperatures and then test empirically using a PCR annealing temperature gradient with gDNA template. LNAs can be used to boost primer annealing temperatures if needed.
Once compatible annealing temperatures have been established, try the multiplex PCR using an equimolar ratio of primers, purify the product, and sequence with shallow long-read sequencing (i.e. Plasmidsaurus Premium PCR sequencing or similar). Check that the sequencing yield of each target is as desired (typically equal, but you may wish to prioritize a specific amplicon for some applications). Tune primer ratios by titration if needed, and reconfirm with sequencing.
Choosing a product length
Target amplicon length is typically 2-10 kb. The lower limit is set to include multiple nucleosome-bound stretches which are important for genomic alignment, and we do not recommend smaller for most applications. The upper limit is more flexible, but constrained by the same challenges as conventional long-range PCR. However, primer specificity can be even more critical because deamination events within the template can create stronger off-target binding sites.
Poly-X stretches
Details on poly-X stretch considerations are being finalized.
C and G in primers
A matter of ongoing discussion is whether to preferentially avoid G bases only or both G and C bases in primer design. SsDddA converts cytosine to uracil bases, so G bases in the primers have the potential for mismatch upon the first PCR cycle. However, on the second cycle, SsDddA-converted uracil bases that were on the 3' end of the PCR-synthesized strand are now A bases at positions complementary to C within the primer. Some lab members have prioritized the first round of amplification by preferentially excluding G. Others argue that an equal C/G ratio in primers is important to maximize efficient amplification on both the first and second cycles, which is important for exponential amplification and anecdotally reduces biases for top vs bottom strands. In practice, the exact choice may not be critical as the majority of DAF-seq template is anticipated to be nucleosome-protected and unmodified at primer binding locations, and DAF-seq primers have been successfully designed with both strategies.
Feedback
DAF-seq is an emergent technology, and we are still improving our primer design methodology. If you have suggestions or collaboration ideas for making this process more robust, please reach out.
Protein Purification
This section covers the expression and purification of the Simiaoa sunii (SsDddA) cytidine deaminase and the DddI inhibitor used in the DAF-seq Protocol.
We recommend storing SsDddA and DddI aliquots at -80 C. Both SsDddA and DddI are stable for at least 5 freeze-thaw cycles.
Addgene
The dual expression plasmid for SsDddA and the expression plasmid for DddI are now avaiable through Addgene!
Need purified protein?
Email absterga@uw.edu for purified SsDddA and DddI protein aliquots.
SsDddA Purification with FPCL Protocol
This protocol for the expression and purification of SsDddA was compiled and optimized by Devaraja G. Mudeppa (Stergachis Lab, Division of Medical Genetics, University of Washington).
Buffers & Reagents
Buffer A
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| NaCl | 600 mM |
| Glycerol | 10% |
| Triton X-100 | 0.1% |
Denaturation Buffer
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| Imidazole | 20 mM |
| NaCl | 500 mM |
| Guanidine HCl (ThermoFisher, Cat# 24110) | 6 M |
| 2-Mercaptoethanol | 5 mM |
Renaturation Buffer
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| NaCl | 500 mM |
| ZnCl₂ | 10 µM |
| 2-Mercaptoethanol | 10 mM |
Buffer B
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 8.0 | 50 mM |
| NaCl | 600 mM |
| Imidazole | 500 mM |
| Glycerol | 10% |
Storage Buffer
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| NaCl | 500 mM |
| ZnCl₂ | 10 µM |
| DTT | 1 mM |
| Glycerol | 10% |
Additional Reagents
- IPTG stock: 1 M in water
- Kanamycin stock: 100 mg/mL in water
Equipment
- Shaker incubator
- Centrifuge capable of up to 40,000 × g
- Sonicator with a 13 mm probe
- FPLC system
- Spectrophotometer
DddA–DddI Complex Expression
- Transform BL21(DE3) with pColDuet-DddA-DddI, plate on LB-kan agar (50 µg/mL). Incubate overnight at 37 °C.
- Inoculate a single colony from the plate into 12 mL LB containing 6 µL of kanamycin stock (100 mg/mL).
- Grow overnight at 37 °C with shaking at 250 rpm.
- Add 10 mL of the overnight culture to 1 L of LB containing 500 µL of kanamycin stock (100 mg/mL).
- Grow at 37 °C with shaking at 250 rpm for approximately 5 hours.
- Measure OD600 and adjust to OD600 ≈ 0.6 if necessary, using fresh LB medium.
- Briefly cool the culture by placing it on ice for a few minutes.
- Add 300 µL of 1 M IPTG to 1 L of culture to achieve a final concentration of 0.3 mM.
- Incubate at 25 °C with shaking at 250 rpm for approximately 18 hours.
- Pellet cells (4000 RCF, 10 min) and store the cell pellet at −80 °C.
DddA Purification
Note: All purification steps were performed at 4 °C. Unless noted, the flow rate was 2 mL/min.
- Thaw the 1 L culture pellet at room temperature for 10 minutes (skip this step if using a fresh pellet).
- Add 35 mL of Buffer A containing two cOmplete Mini EDTA-free protease inhibitor tablets (Millipore-Sigma; Cat# 11836170001).
- Vortex vigorously for 2–3 minutes.
- Transfer the resuspended cells to a 50 mL Falcon tube and vortex again to eliminate lumps.
- Sonicate with a 13 mm probe for 5 minutes (30 s on/30 s off cycles) at 30% amplitude; total sonication time is 10 min.
- Centrifuge the lysate at 40,000 × g for 1 hour.
- Load the supernatant onto a 5 mL HisTrap Excel column (Cytiva; Cat# 17371206), which has been pre-equilibrated with 5 column volumes (CV) of Buffer A.
- Wash the column with 5 CV of Buffer A, then 5 CV Renaturation Buffer.
- Denature column-bound DddA/DddI by applying a 10 CV gradient from 100% Renaturation Buffer to 100% Denaturation Buffer.
- Maintain the column in Denaturation Buffer for an additional 5 CV.
- Renature DddA by applying a 15 CV gradient from 100% Denaturation Buffer to 100% Renaturation Buffer.
- Maintain the column in Renaturation Buffer for an additional 10 CV at 1 mL/min.
- Elute DddA using 5 CV of Buffer B.
- Pool protein-containing fractions (based on A280) and concentrate to 1 mL using a 15 mL 3 kDa MWCO Amicon filter (Millipore Sigma; Cat# UFC900308).
- Perform buffer exchange by diluting the protein to 15 mL with Storage Buffer and reconcentrating to 1 mL; repeat this step 3 times.
- Quantify purified DddA using the Bradford assay. Adjust DddA concentration to 100 µM using Storage Buffer. Verify purity using 4–20% SDS-PAGE.
- Store the purified DddA in required aliquots at −80 °C. The DddA is stable for at least five cycles of freeze-thaw.
DddI Purification with FPLC Protocol
This protocol for the expression and purification of DddI was compiled and optimized by Devaraja G. Mudeppa (Stergachis Lab, Division of Medical Genetics, University of Washington).
Buffers & Reagents
Buffer A
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| NaCl | 600 mM |
| Glycerol | 10% |
| Imidazole | 10 mM |
| Triton X-100 | 0.1% |
| 2-Mercaptoethanol | 5 mM |
Wash Buffer
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| NaCl | 500 mM |
| Imidazole | 30 mM |
| 2-Mercaptoethanol | 5 mM |
Buffer B
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 8.0 | 50 mM |
| NaCl | 600 mM |
| Imidazole | 500 mM |
| Glycerol | 10% |
Storage Buffer
| Component | Final Concentration |
|---|---|
| Tris-HCl, pH 7.8 | 50 mM |
| NaCl | 500 mM |
| DTT | 1 mM |
| Glycerol | 10% |
Additional Reagents
- IPTG stock: 1 M in water
- Kanamycin stock: 100 mg/mL in water
Equipment
- Shaker incubator
- Centrifuge capable of up to 40,000 × g
- Sonicator with a 13 mm probe
- FPLC system
- Spectrophotometer
DddI Expression
- Transform BL21(DE3) with pColDuet_nHis_SsDddI, plate on LB-kan agar (50 µg/mL). Incubate overnight at 37 °C.
- Inoculate a single colony from the plate into 12 mL LB containing 6 µL of kanamycin stock (100 mg/mL).
- Grow overnight at 37 °C with shaking at 250 rpm.
- Add 10 mL of the overnight culture to 1 L of LB containing 500 µL of kanamycin stock (100 mg/mL).
- Grow at 37 °C with shaking at 250 rpm for approximately 5 hours.
- Measure OD600 and adjust to OD600 ≈ 0.6 if necessary, using fresh LB medium.
- Briefly cool the culture by placing it on ice for a few minutes.
- Add 300 µL of 1 M IPTG to 1 L of culture to achieve a final concentration of 0.3 mM.
- Incubate at 25 °C with shaking at 250 rpm for approximately 18 hours.
- Pellet cells (4000 × g, 10 min) and store the cell pellet at −80 °C.
DddI Purification
Note: All purification steps were performed at 4 °C. Unless noted, the flow rate was 2 mL/min.
- Thaw the 1 L culture pellet at room temperature for 10 minutes (skip this step if using a fresh pellet).
- Add 35 mL of Buffer A containing two cOmplete Mini EDTA-free protease inhibitor tablets (Millipore-Sigma; Cat# 11836170001). Vortex vigorously for 2–3 minutes.
- Transfer the resuspended cells to a 50 mL Falcon tube and vortex again to eliminate lumps.
- Sonicate with a 13 mm probe for a total time of 10 min (30 s on/30 s off cycles) at 30% amplitude.
- Centrifuge the lysate at 30,000 × g for 30 min.
- Load the supernatant onto a 5 mL HisTrap Excel column (Cytiva; Cat# 17371206), which has been pre-equilibrated with 5 column volumes (CV) of Buffer A.
- Wash the column with 5 CV of Buffer A, then 10 CV Wash Buffer.
- Elute column-bound DddI by applying a 5 CV (25 mL) gradient from 100% Wash Buffer to 100% Buffer B.
- The protein eluted between ~30% and ~70% was collected.
- Concentrate eluted DddI using a 15 mL 3 kDa MWCO Amicon filter (Millipore Sigma; Cat# UFC900308).
- Perform buffer exchange by diluting the protein to 15 mL with Storage Buffer and reconcentrating to 1 mL; repeat this step 3 times.
- Quantify purified DddI using the Bradford assay. Adjust DddI concentration to 1000 µM using Storage Buffer. Verify purity using 4–20% SDS-PAGE.
- Store the purified DddI in required aliquots at −80 °C. The DddI is stable for at least 5 freeze-thaw cycles.
DAF-QC Pipeline
DAF-QC-SMK is a Snakemake pipeline for quality control and initial processing of DAF-seq sequencing reads. It supports both PacBio HiFi and Oxford Nanopore platforms. This page covers installation, usage, and key outputs. For the wet lab steps that precede this pipeline, see the DAF-seq Protocol.
Getting started
The pipeline uses pixi for environment management. Clone the repository and install:
git clone https://github.com/StergachisLab/DAF-QC-SMK.git
cd DAF-QC-SMK
pixi install
Verify the installation
A test dataset (human chr8, hg38) is bundled with the repository. Run it to confirm everything is working before processing your own data:
pixi run test
If you encounter errors, please run the test case before contacting the developers, as it helps with troubleshooting.
Usage
Run the pipeline with pixi:
pixi run snakemake --configfile config/config.yaml
For SLURM clusters, specify a profile:
pixi run snakemake --configfile config/config.yaml --profile profiles/slurm-executor
You can also run the pipeline from a different directory using --manifest-path:
pixi run --manifest-path /path/to/DAF-QC-SMK/pixi.toml snakemake --configfile config/config.yaml
Inputs
The pipeline requires two configuration files:
Sample table (config.tbl)
A tab-separated table with sample name, BAM/FASTQ path, and targeted regions:
sample file regs
test test.bam chr8:144415767-144417958
For PacBio BAM inputs, files should contain either unaligned reads or primary reads only (for compatibility with pbmarkdup during consensus generation). See config/config.tbl in the repository for a template.
Configuration file (config.yaml)
Specifies paths to the sample table and reference genome, sequencing platform, and optional parameters:
ref: /path/to/genome.fa
manifest: config/config.tbl
platform: pacbio # 'pacbio' or 'ont'
# Optional (both platforms)
chimera_cutoff: 0.9
min_deamination_count: 50
end_tolerance: 30
decorated_samplesize: 5000
# PacBio-specific
consensus: True
consensus_min_reads: 3
# ONT-specific
is_fastq: False
See config/config.yaml in the repository for the full list of options with descriptions.
Key outputs
- Aligned BAMs: Primary, supplementary, and unaligned reads with PCR duplicates marked (
duanddstags). - Decorated BAMs: Full-length reads with top/bottom strand designation (C-to-T as top strand, G-to-A as bottom strand). Strand stored in the
sttag. - Consensus BAMs (PacBio only): MSA consensus of full-length, strand-designated reads. The
dctag indicates the number of reads used to construct each consensus. - QC metrics: Targeting efficiency, deamination rates (overall and by 2-bp sequence context), strand calling, enzyme bias, and mutation rates.
- HTML dashboard:
results/{sample_name}/qc/{sample_name}.dashboard.htmlwith all QC plots. The dashboard is self-contained (plots are embedded), so you can copy a single file for sharing or local viewing.
Downstream analysis
After QC, DAF-seq data can be processed for nucleosome, MSP, and transcription factor footprint calling with FiberHMM, a Hidden Markov Model toolkit that operates natively on deaminase data (DddA and DddB) and emits fibertools-compatible BAMs plus Molecular-annotation spec tags. See the FiberHMM page for installation and usage.
Alternatively, the Fiber-seq nucleosome caller ft add-nucleosomes can be applied to DAF-seq data after first converting the deamination marks to m6A-equivalent format with ft ddda-to-m6a. This routes DAF-seq data through the Fiber-seq analysis stack; see the fibertools documentation for details.
Further reading
See the DAF-QC-SMK README for full details on all configuration options, output file formats, and BAM tag specifications.
FiberHMM
FiberHMM is a Hidden Markov Model toolkit for calling chromatin footprints from single-molecule DNA modification data. It supports DAF-seq (DddA and DddB) as well as Fiber-seq (PacBio and Nanopore Hia5), and emits nucleosomes, methylase-sensitive patches (MSPs), and sub-nucleosomal TF/Pol II footprints in fibertools-compatible BAMs.
For DAF-seq, FiberHMM is a native nucleosome and footprint caller that runs directly on deaminase data with DAF-trained HMM emissions, adds a log-likelihood-ratio recaller for transcription factor footprints, and writes spec-compliant Molecular-annotation tags. This page covers installation, inputs, and the recommended one-command workflow.
Getting started
Install from PyPI:
pip install fiberhmm
Optional dependencies that are worth installing:
pip install numba # ~10x faster HMM computation
pip install matplotlib # --stats visualization
pip install h5py # HDF5 posteriors export
For bigBed output, install bedToBigBed from UCSC tools.
Pre-trained models for DddA and DddB are bundled with the package; no separate download is required.
Inputs
FiberHMM operates on aligned DAF-seq BAMs from the DAF-QC pipeline (or any equivalent alignment workflow). In DAF mode, the caller needs to know which positions on each read are C-to-T or G-to-A conversions. It auto-detects this per read from any of:
- R/Y IUPAC codes in the stored sequence, written by
fiberhmm-daf-encode. - MD tag on a raw aligned BAM (produced by
minimap2 --MDorsamtools calmd). Parsed on the fly; no preprocessing step needed. - MM/ML tags encoding deaminated C/G positions as base modifications.
--reference ref.fa, used as a fallback when none of the above are present.
At least one of these must be present on the input BAM.
Usage
fiberhmm-call is the recommended entry point. It fuses the nucleosome/MSP HMM and the TF recaller into a single in-process pipeline.
DddA
FiberHMM automatically selects the DddA two-model workflow under the hood (a nucleosome model plus a TF recall pass with an efficiency uplift to match DddA's higher per-position deamination rate).
fiberhmm-call -i aligned.bam -o recalled.bam \
--mode daf --enzyme ddda \
-c 8 --io-threads 16 \
--region-parallel
--region-parallel requires a coordinate-sorted and indexed input and scales near-linearly with --cores up to the chromosome count. The output is sorted and indexed in place; no separate sort pass is needed.
DddB samples are supported by the same commands with --enzyme dddb.
Key outputs
fiberhmm-call writes a tagged BAM that downstream tools like FiberBrowser and Fibertools can read directly.
- Legacy footprint tags (
ns/nl,as/al): nucleosome and MSP starts and lengths, compatible with any tool in the fibertools ecosystem. - Molecular-annotation spec tags (
MA,AQ): per-specnuc+Q,msp+, andtf+QQQannotations with LLR-based confidence (tq) and edge-sharpness bytes (el,er) on TF calls. See the spec for the encoding. - TF/Pol II footprints: sub-nucleosomal calls (typically 15–80 bp) live in the
tf+QQQannotation ofMA/AQ. For DAF-seq, the recaller uses a tuned--min-llr(5.0 for DddA, 4.0 for DddB) selected automatically by--enzyme.
Extract to bigBed
For a smaller-filesize representation of the call set for downstream analysis and FiberBrowser, convert the tagged BAM:
fiberhmm-extract -i recalled.bam --footprint --msp --tf --bigbed
Choosing options
| Situation | Command |
|---|---|
| Default: full pipeline on a sorted + indexed DAF-seq BAM | fiberhmm-call --mode daf --enzyme ddda --region-parallel |
| Want FIRE element calls afterwards | Pipe fiberhmm-call -o - into ft fire - final.bam |
| DddB samples | swap --enzyme ddda for --enzyme dddb in any of the above |
For Hia5 fiber-seq usage and the full CLI surface (training new models, exporting posteriors, model inspection), see the FiberHMM README.
Further reading
- FiberHMM repository and README
- Molecular-annotation spec for the
MA/AQtag schema - fibertools-rs for downstream FIRE scoring and extraction
FiberBrowser
FiberBrowser is an interactive, single-molecule-genomics-focused genome browser for DAF-seq, Fiber-seq, and a variety of other genomic datatypes. It is the visualization component of the FiberHMM pipeline, but does not require footprints to be useful. It allows fast and convenient exploration and quantitative analysis of per-read footprinting and methylation/deamination patterns across 1000s of reads, providing extensive functionality missing from modern browsers which focus on aggregate short-read tracks.
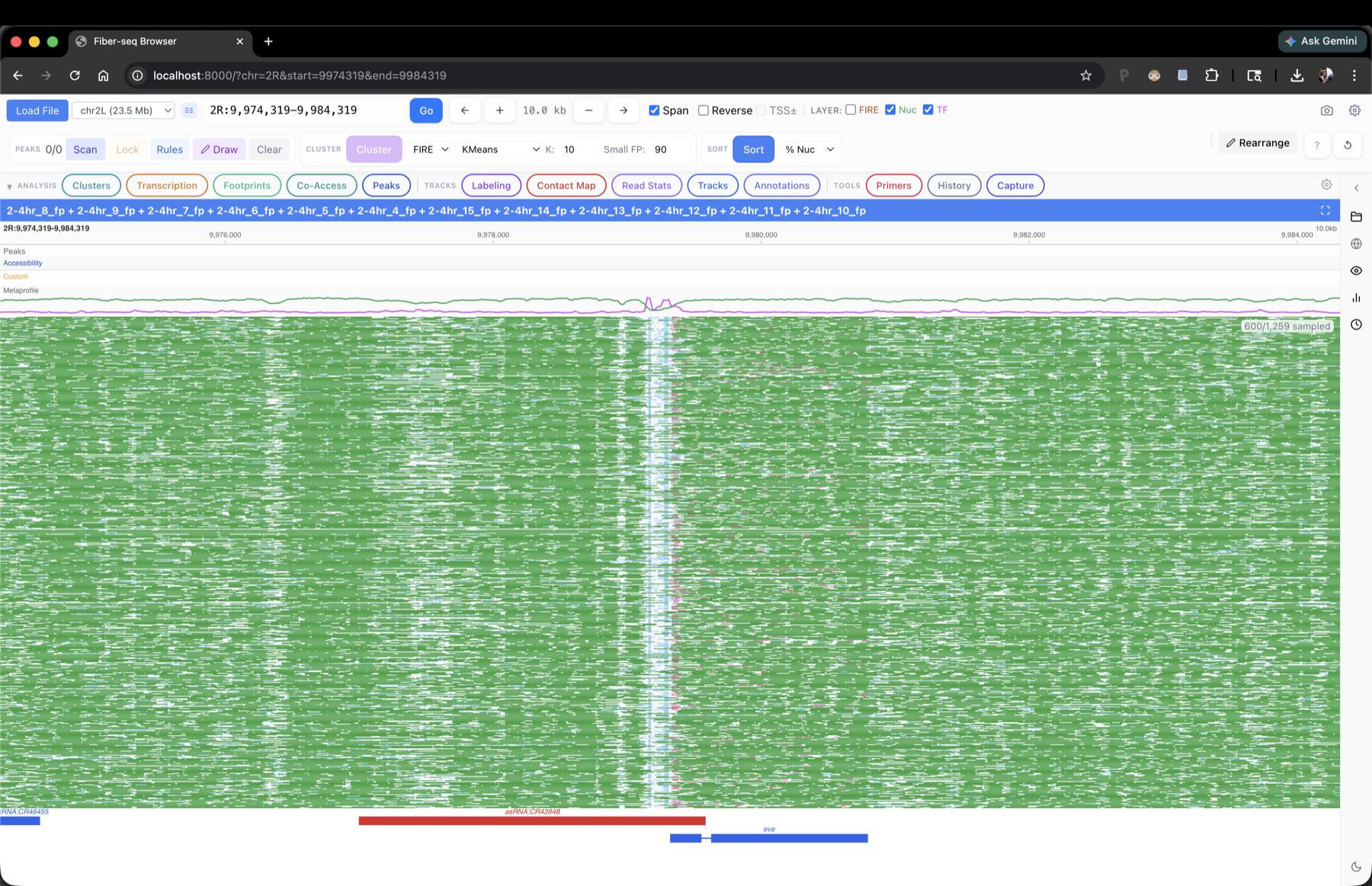
2-4 hr Fiber-seq at the eve locus in Drosophila embryos. 1,259 reads span the region; the browser draws every footprint on every read, and every downstream analysis is computed on the full read set.
Features
- Per-read heatmaps of m6A / DAF accessibility, drawn as a single canvas layer so loci with thousands of reads stay responsive.
- Single-molecule clustering on CRE accessibility patterns, with per-cluster metaprofiles, merge / reorder / hide controls, and a sortable read table.
- Direct comparison across perturbations or timepoints -- load multiple datasets at once and joint-cluster them so the same single-molecule state definitions apply across conditions, making perturbation effects readable at single-read resolution.
- Customizable footprint labeling -- recolor footprints by user-defined rules (Pol II, NFR, TF, nucleosome) using size and overlap criteria.
- Transcription-state quantification -- filter and count reads matching paused, PIC, elongating, or hyperbound Pol II configurations at a selected gene.
- Footprint inspection and motif enrichment -- pull motif hits and FASTA for any single footprint and compare to references (ChIP-nexus, motif databases).
- DAF-seq primer design -- design and BLAST-check primers for targeted DAF-seq amplicons directly from the browser, with IDT / Twist CSV export. See also the Primer Design guidelines.
- Headless / API mode and Jupyter control -- drive the browser programmatically via its FastAPI endpoints or a Python client; load regions, pull cluster assignments, fetch co-accessibility matrices, and render figures directly from a notebook.
- Local LLM copilot (optional) -- natural-language commands that drive the browser via a model running on your machine (MLX on Mac, Ollama elsewhere); doubles as an interactive tutorial for discovering features.
- ...and more.
FiberBrowser sits downstream of the DAF-QC Pipeline: once you have QC'd reads and called footprints, FiberBrowser is where you explore them.
Early access
FiberBrowser is in pre-release while we finalize the public repository. If you'd like to try it on your data, reach out at tommywtullius [at] gmail.com.
Glossary
DAF-seq (Deaminase-Assisted single-molecule chromatin Fiber sequencing): A method for single-molecule footprinting at near-nucleotide resolution that simultaneously profiles chromatin states and DNA sequence. It uses cytosine deamination on intact nuclei to stencil protein occupancy along individual DNA fibers.
SsDddA: A nonspecific double-stranded DNA cytidine deaminase from Simiaoa sunii. In DAF-seq, SsDddA is applied to intact nuclei where it deaminates accessible (non-protein-bound) cytosines to uracils, which appear as C-to-T transitions after sequencing.
DddI: A protein inhibitor of DddA-family deaminases. In DAF-seq, DddI is added at a 5-fold molar excess over SsDddA to stop the deamination reaction.
UNG inhibitor: Uracil-N-glycosylase inhibitor (UGI). Added before the deamination step to prevent endogenous UNG from removing uracils created by SsDddA, which would cause DNA strand breaks and data loss.
scDAF-seq: Single-cell DAF-seq. A variant of DAF-seq that uses primary template-directed amplification (PTA) to enable single-cell chromatin fiber profiling.
Decorated BAM: A BAM file in which full-length DAF-seq reads have been assigned a top or bottom strand designation based on deamination patterns (C-to-T for top strand, G-to-A for bottom strand). Strand designation is stored in the st BAM tag.
Chimera: A sequencing read that contains deamination signal from both strands, indicated by a mixture of C-to-T and G-to-A conversions. Chimeric reads are filtered using a configurable cutoff (default: 90% single-strand signal).
Full-length read: A read that spans the entire targeted amplicon (within a configurable end tolerance, default 30 bp). Only full-length reads are used for strand designation and downstream analysis.
Consensus BAM (PacBio only): A BAM file containing multiple-sequence-alignment (MSA) consensus sequences built from groups of PCR duplicate reads. Consensus generation improves accuracy of deamination calls.
fibertools: A suite of command-line tools for analyzing single-molecule chromatin fiber data. The ddda-to-m6a subcommand converts DAF-seq deamination marks (i.e. Y and R bases) into m6A-equivalent format for compatibility with Fiber-seq analysis tools, and add-nucleosomes infers nucleosome positions from the resulting data.
Cite
If you use DAF-seq in your research, please cite the following:
Swanson, E. G., Mao, Y., Mallory, B. J., Vollger, M. R., Bohaczuk, S. C., Oliveira, C. B., Lyon, D. B., Ranchalis, J., Parmalee, N. L., Cohen, B. A., Bennett, J. T. & Stergachis, A. B. Mapping single-cell diploid chromatin fiber architectures using DAF-seq. Nature Biotechnology (2025). https://doi.org/10.1038/s41587-025-02914-3
BibTeX
@article{swanson2025dafseq,
title = {Mapping single-cell diploid chromatin fiber architectures using {DAF}-seq},
author = {Swanson, Elliott G. and Mao, Yizi and Mallory, Benjamin J. and
Vollger, Mitchell R. and Bohaczuk, Stephanie C. and
Oliveira, Christopher B. and Lyon, Daniel B. and Ranchalis, Jane and
Parmalee, Nancy L. and Cohen, Barak A. and Bennett, James T. and
Stergachis, Andrew B.},
journal = {Nature Biotechnology},
year = {2025},
doi = {10.1038/s41587-025-02914-3},
url = {https://doi.org/10.1038/s41587-025-02914-3}
}
Related resources
- DAF-seq Manuscript repository -- Analysis code and supplementary materials
- DAF-QC-SMK -- QC pipeline for DAF-seq data